Subscribe for E-mail Alerts
Subscribe for E-mail Alerts CCDC Weekly RSS Feed
CCDC Weekly RSS Feed2022 Vol. 4, No. 12

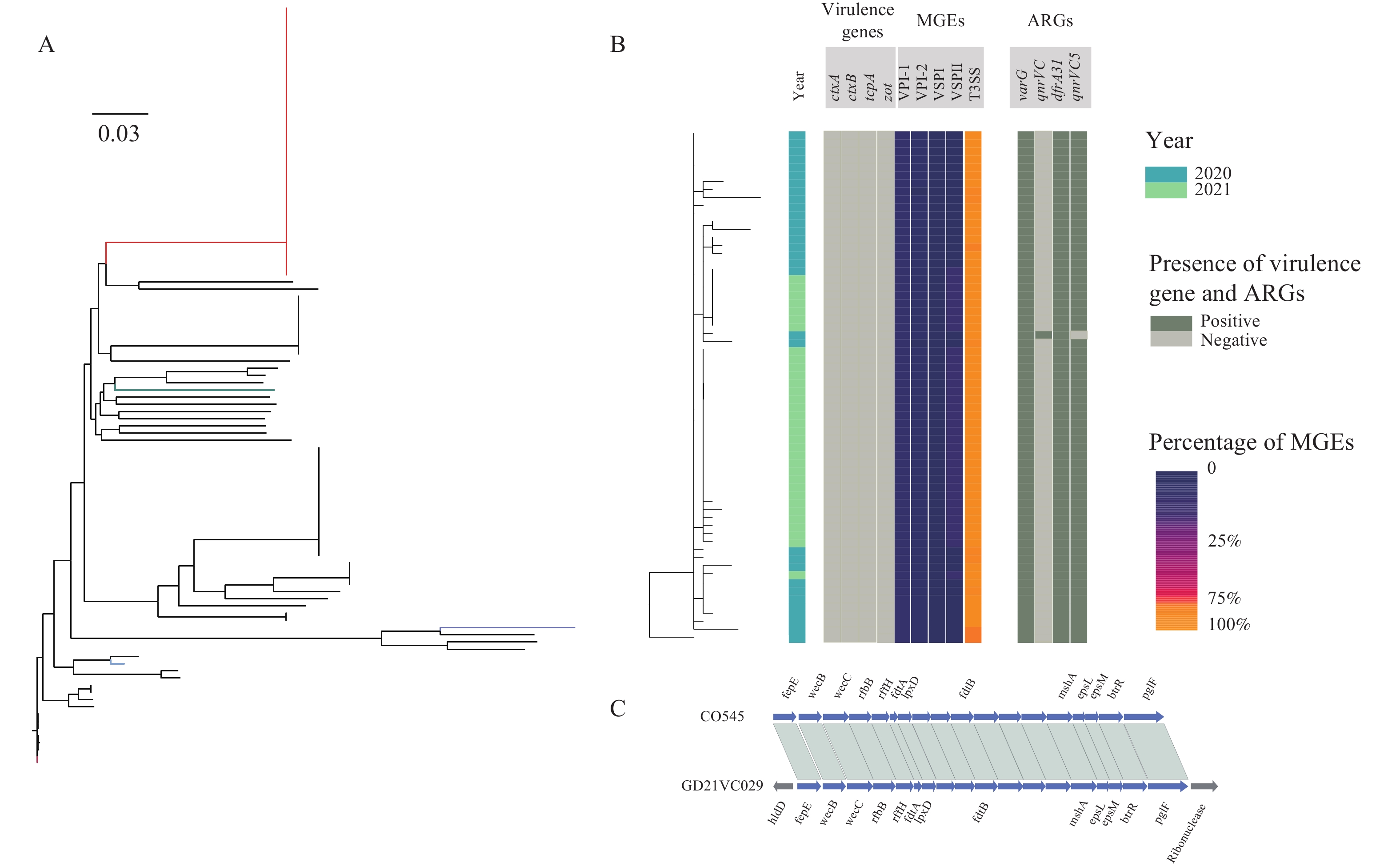
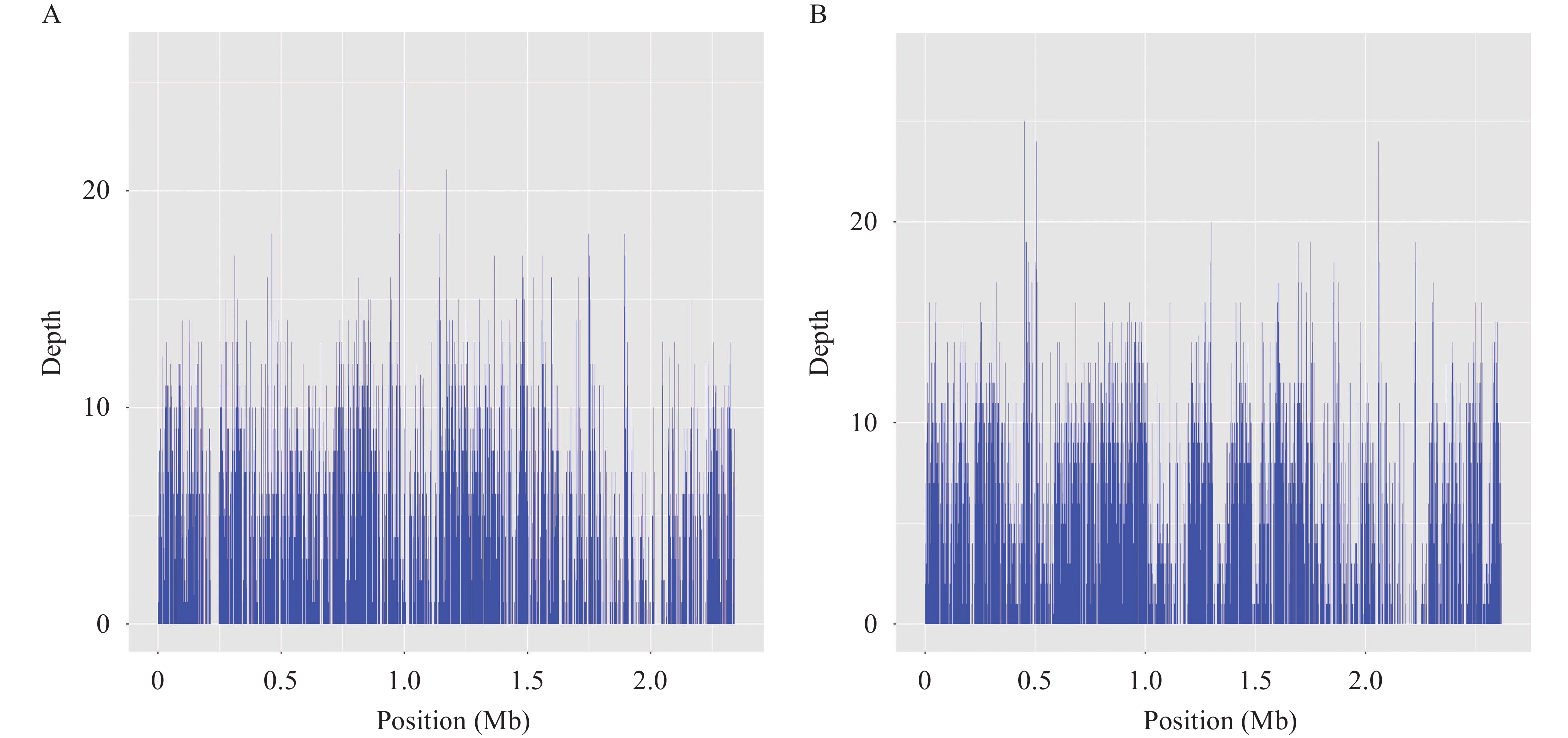
Gastroenteritis caused by non-O1/non-O139 Vibrio cholerae exhibited an increasing trend in recent years in China. Whole genome sequence (WGS) data could play an important role both in the identification of the outbreaks and in the determination of the serogroup. Here, we present the employment of WGS data in the investigation of two outbreaks caused by non-O1/non-O139 V. cholerae in Guangdong, China, 2020–2021.
We obtained the whole genome sequence of 66 V. cholerae strains isolated in two outbreaks with next generation sequencing technology. We retrieved the publicly available WGS data of non-O1/non-O139 V. cholerae from public database. We used a pipeline integrated in China Pathogen Identification Net (PIN) to complete the phylogenetic analysis.
Two outbreaks caused by non-O1/non-O139 V. cholerae were identified using WGS data. These V. cholerae strains were determined as serogroup O5. Type 3 and 6 secretion systems were detected in these serogroup O5 strains. These serogroup O5 strains belonged to sequence type (ST) 88.
Our analysis indicated the risk of non-O1/non-O139 V. cholerae leading to outbreaks of diarrheal diseases. The application of genomic data played an important role in the identification of the serogroup of non-O1/non-O139 V. cholerae in the lack of antiserum, which gave an example of the application of genome data in disease surveillance and public health emergency response.

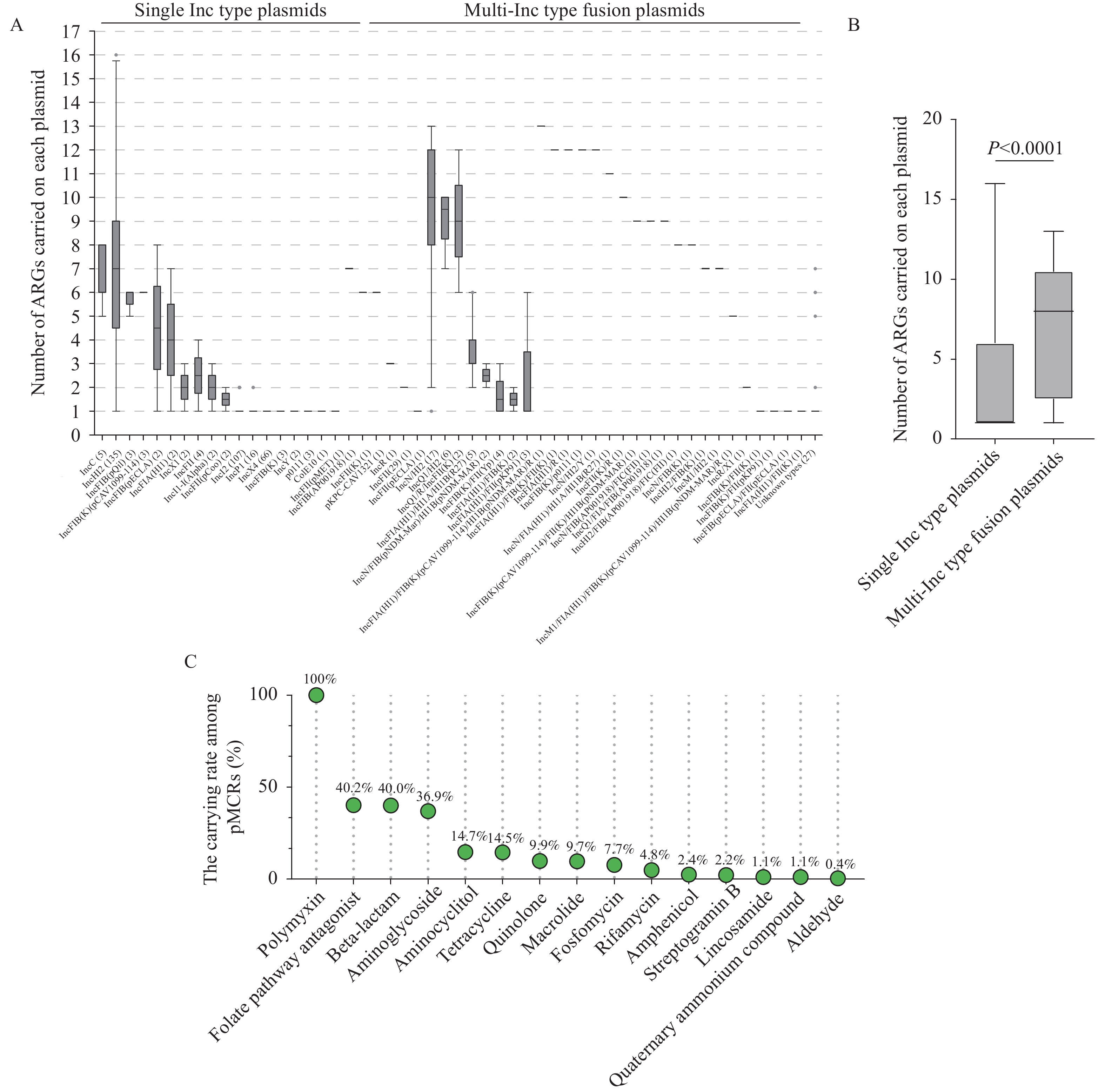
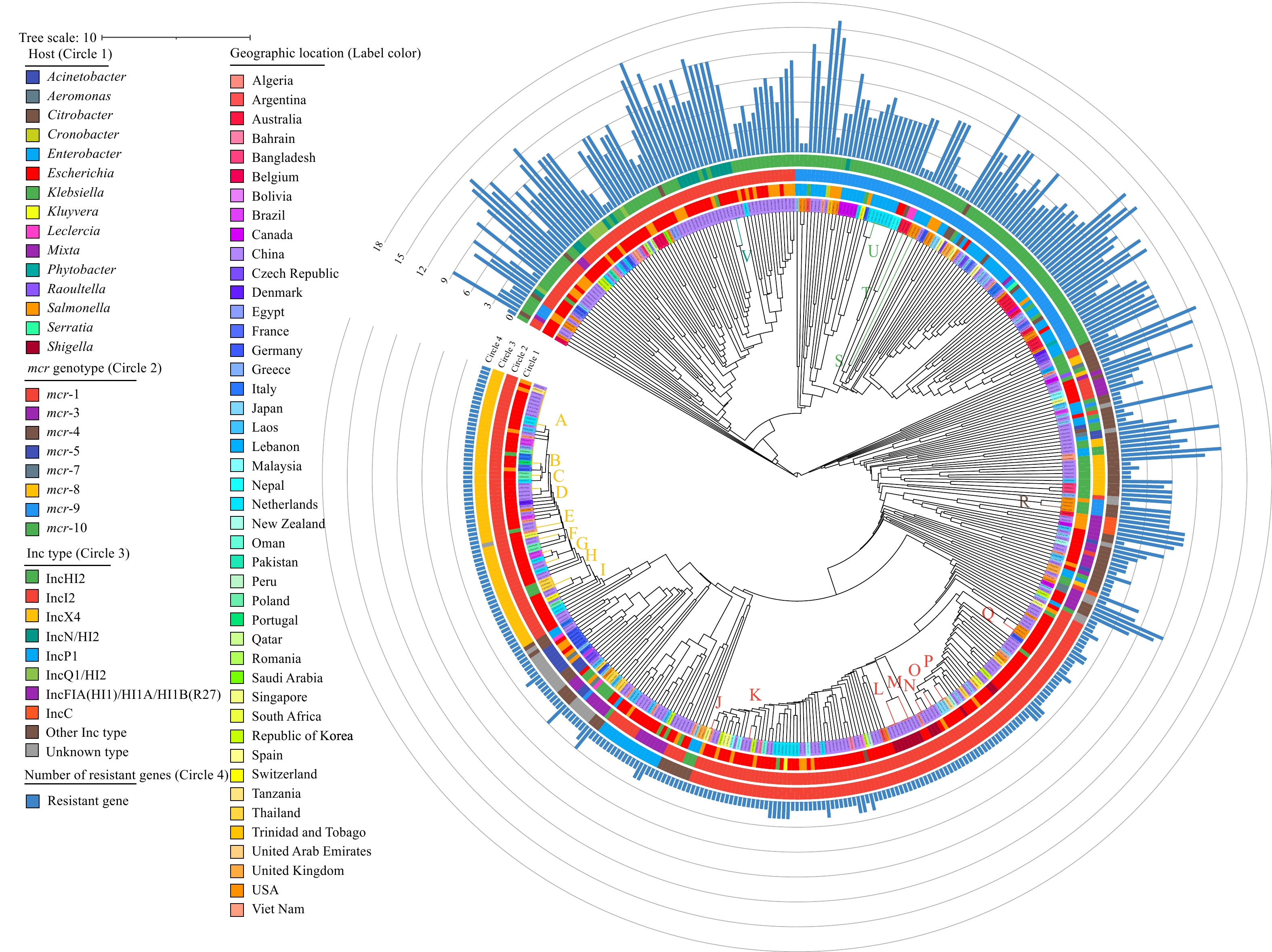
The surveillance of antimicrobial resistance genes (ARGs) and bacteria is one critical approach to prevent and control antimicrobial resistance (AMR). Next-generation sequencing (NGS) is a powerful tool in monitoring the emergence and spread of ARGs and resistant bacteria. The horizontal transfer of ARGs across host bacteria mediated by plasmids is a challenge in NGS surveillance for resistance because short-read sequencing can hardly generate the complete plasmid genome sequence, and the correlation between ARGs and plasmids are difficult to determine.
The complete genome sequences of 455 mcr-carrying plasmids (pMCRs), and the data of their host bacteria and isolation regions were collected from the NCBI database. Genes of Inc types and ARGs were searched for each plasmid. The genome similarity of these plasmids was analyzed by pangenome clustering and genome alignment.
A total of 52 Inc types, including a variety of fusion plasmids containing 2 or more Inc types were identified in these pMCRs and carried by complex host bacteria. The cooccurrence of ARGs in pMCRs was generally observed, with an average of 3.9 ARGs per plasmid. Twenty-two clusters with consistent or highly similar sequences and gene compositions were identified by the pangenome clustering, which were characterized with distributions in different countries/regions, years or host bacteria in each cluster.
Based on the complete plasmid sequences, distribution of mcr genes in different Inc type plasmids, their co-existence with other AMRs, and transmission of one pMCR across regions and host bacteria can be revealed definitively. Complete plasmid genomes and comparisons in the laboratory network are necessary for spread tracing of ARG-carrying plasmids and risk assessment in AMR surveillance.

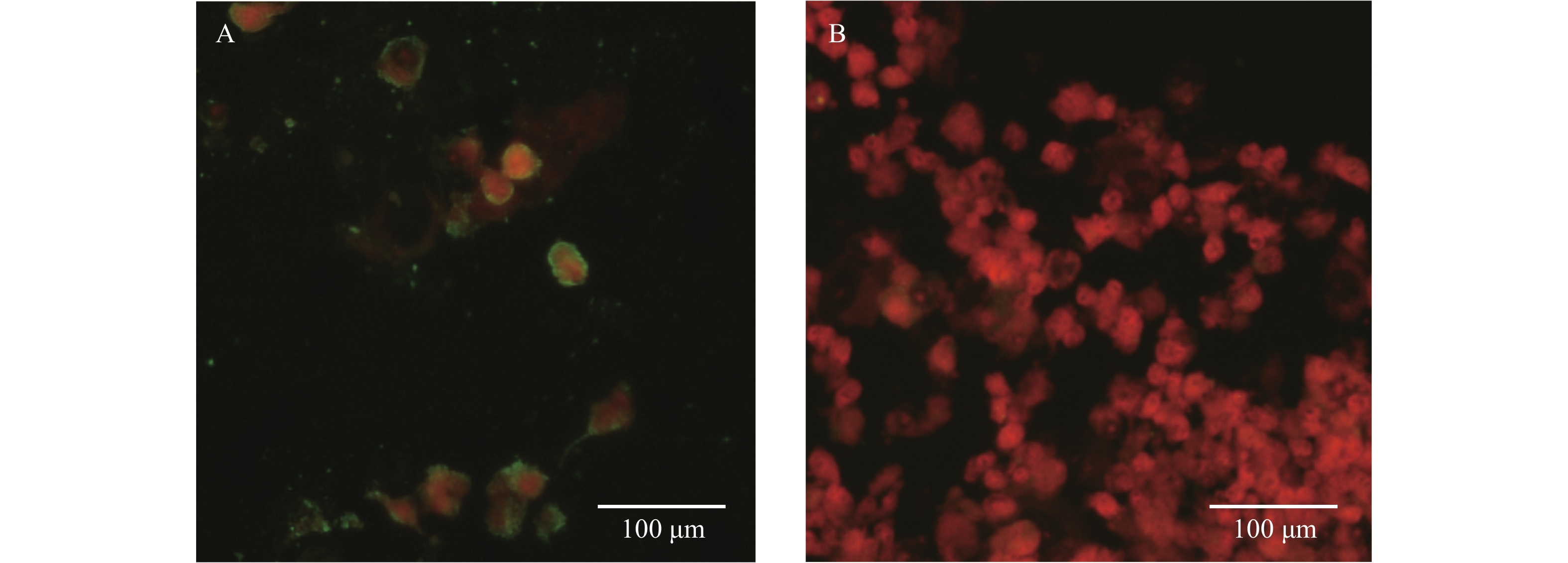
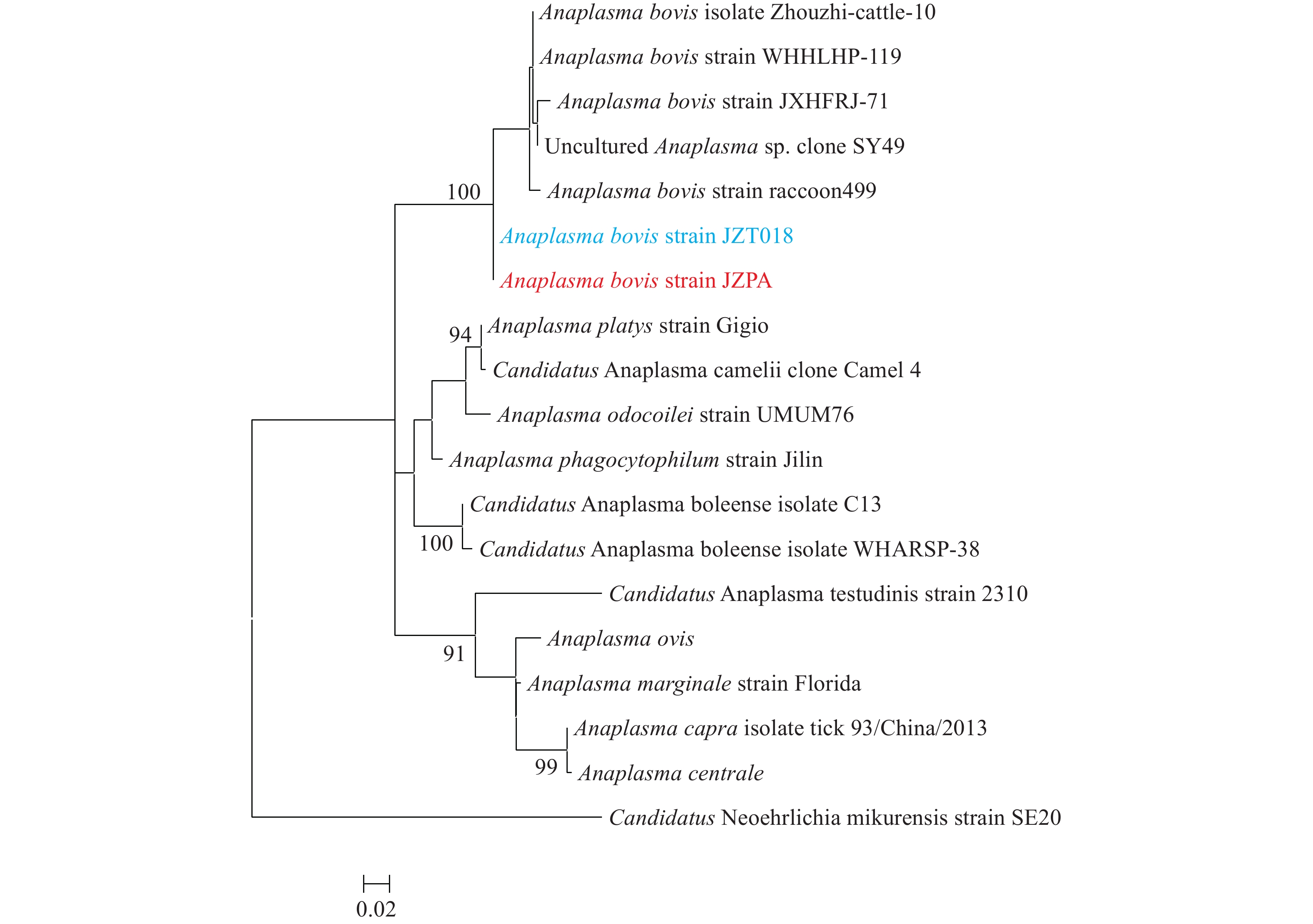
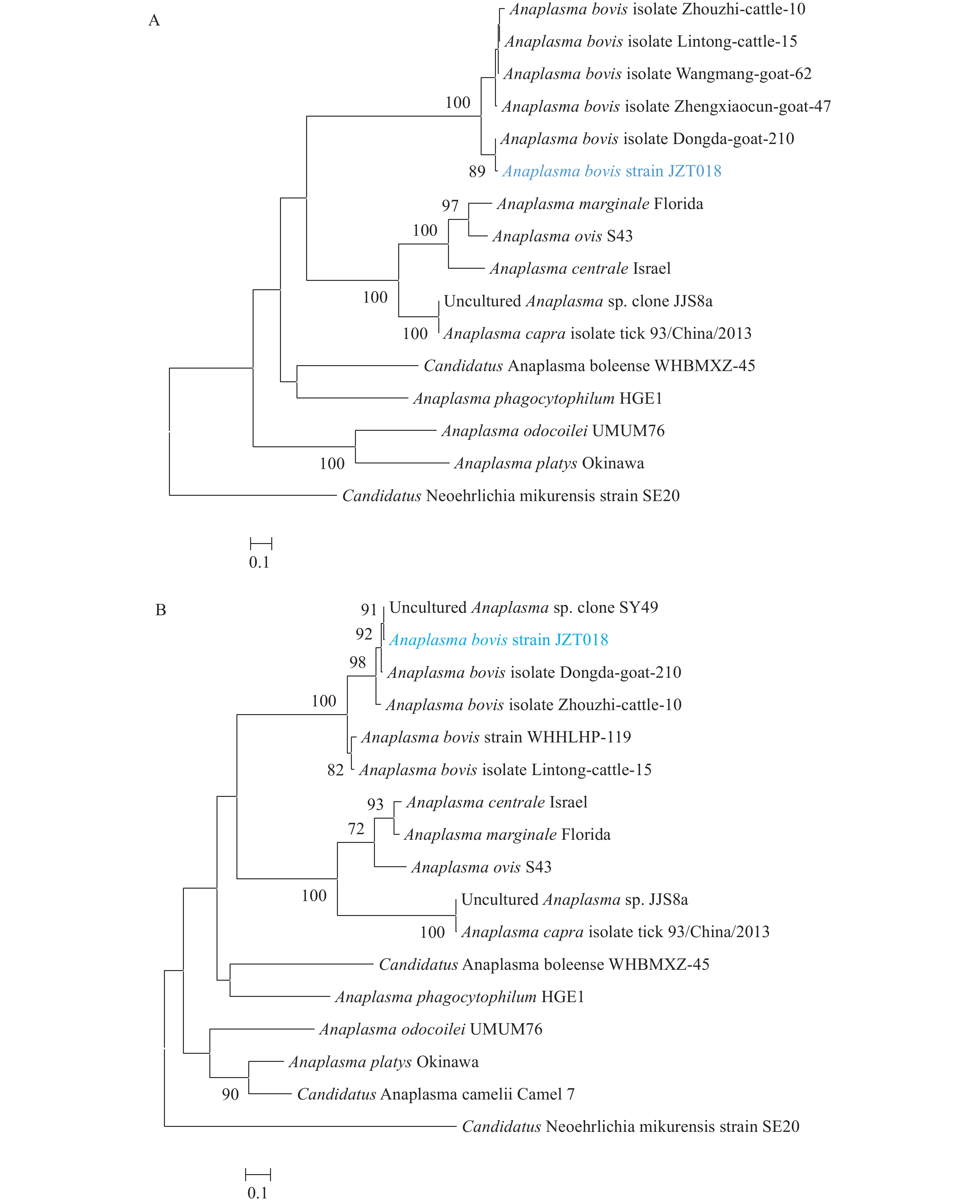
The genus Anaplasma contains seven recognized bacterial species, mainly transmitted by tick bites. The two species, A. phagocytophilum and A. capra, are known commonly to cause diseases in humans.
Anaplasma bovis was initially thought to be only an animal agent until the first patient case was reported in 2019. This study investigated another two patients who became sick within one month in the same township and were infected with A. bovis in Anhui Province.
This study suggested that more A. bovis-infected patients may exist in this area and that patients with anaplasmosis require an early and specific diagnosis.
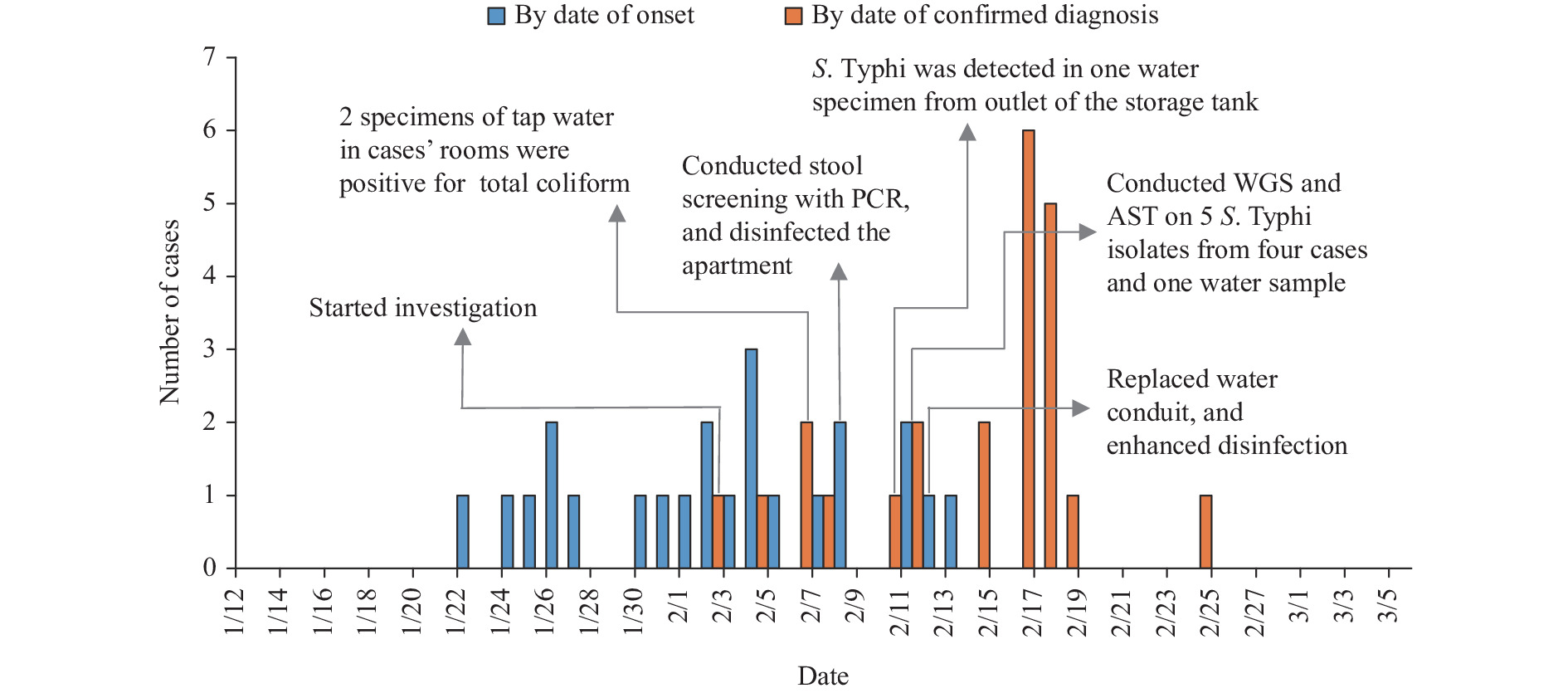
Typhoid fever remains a major public health problem in developing countries. Waterborne typhoid fever affects an estimated 27 million people worldwide each year. Decades of indiscriminate antibiotic usage has driven the emergence and spread of multidrug-resistant (MDR) and even extensively drug-resistant (XDR) Salmonella Typhi (S. Typhi) strains.
By combining the epidemiological investigations, bacterial isolation from patients and household water, whole genome sequencing and drug resistance analysis, we identified a waterborne typhoid fever outbreak caused by XDR S. Typhi in Beijing municipality. This was the first report of the XDR S. Typhi triggered outbreak in Beijing, which was also rare in China.
This report highlights the importance of ensuring access to affordable and safe drinking water, improved sanitation, and waste management systems for resource-constrained urban populations. Typhoid fever caused by XDR S. Typhi is still a severe public health threat.

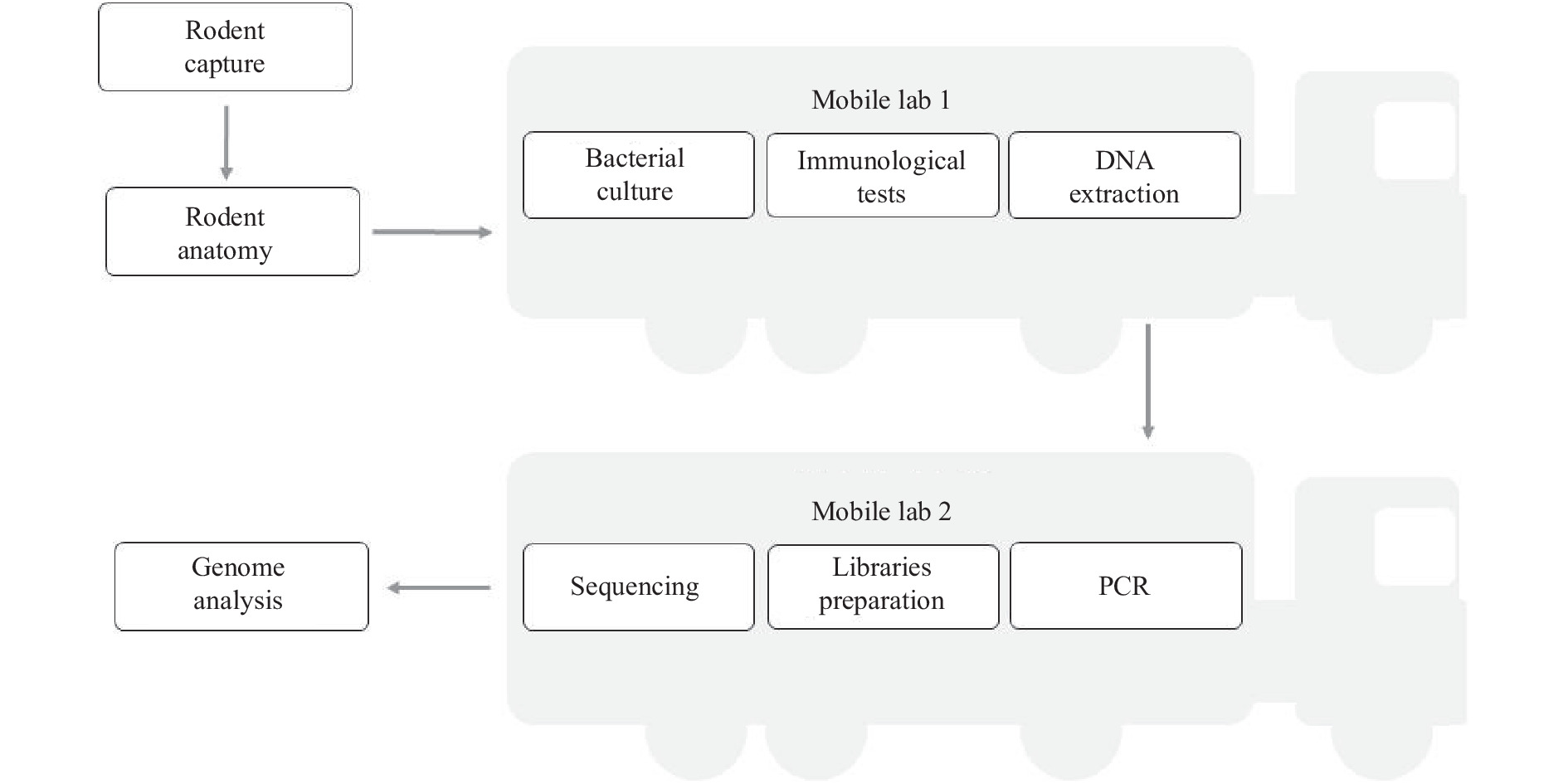
Accurate etiological detection is needed to evaluate the risk of zoonotic diseases. Metagenomic next-generation sequencing (mNGS) can be used to monitor pathogens in animal species and identify potential zoonotic threats. The current sampling model for zoonotic pathogen monitoring in wild animals requires samples to be transferred from the field to a laboratory for further detection.
We constructed a zoonotic pathogen survey model using a set of mobile laboratories.
The monitoring in this study was preplanned to detect Yersinia pestis, but the mNGS unexpectedly identified Bartonella spp. in the rodent samples, thus exposing the threat of bartonellosis to humans in this region. The co-localization of sampling and sequencing (CLOSS) model we tested required no long-distance transferring of samples and expands the regional coverage of zoonotic surveys by using a mobile laboratory.
Using this mNGS technique will enable detection of more zoonotic pathogens beyond the preplanned monitoring targets. This may increase the surveillance efficiency compared with that of the previous workflow and expand the application of the mobile laboratories for infectious diseases identification and surveillance in the field.