
 Download:
Download:




-
The misuse of antimicrobials in clinical and veterinary medicine for prophylactic, therapeutic, and growth promoters (1-2) accelerates the emergence and spread of antimicrobial resistance, posing a threat to the effective control of bacterial diseases. Coupled with the cross regional speed of resistance caused by the acceleration of globalization, a long-term mechanism of global surveillance for antimicrobial resistance genes (ARGs), such as a laboratory network, should be established. ARGs spread among bacteria through vertical transmission and horizontal transfer, in which plasmid-mediating ARG transmission is quite active and even spread across bacterial species. Next-generation sequencing (NGS) of microbial genomes has been widely applied in the laboratory and epidemiological surveillance for infectious diseases. Because of the limitations of short-read sequencing techniques, complete plasmid sequences are hard to be assembled from short reads, which results in the data loss of plasmid-carrying genes and difficulty in determining if ARGs are carried by plasmids. Therefore, the tracing of resistant plasmids will be unable to implement in the resistance surveillance.
The colistin was reintroduced as a last resort in the treatment of carbapenem-resistant Enterobacteriaceae infections (3). However, the plasmid-mediated colistin resistance gene (mcr) was found in 2015, which increases the threat of rapid transmission of colistin resistance (4). In the following years, 10 genotypes of mcr genes have been reported in more than 50 countries on 6 continents (5), and their host bacteria were isolated from animals, environment, and humans (6). Besides mcr genes, monitoring and tracing of their plasmid vectors are also necessary in understanding, spread assessment, and control of mcr-mediated resistance.
In this study, we retrieved the sequences of mcr-carrying plasmids (pMCRs) which have the complete plasmid genomes and analyzed their resistance gene contents and genome clustering. Possible transmission of pMCRs was found across countries, years, and hosts, which revealed the important roles of the complete plasmid genomes for the tracing of resistant plasmids in antimicrobial resistance surveillance.
-
A total of 455 pMCRs along with their geographic locations, collection years, and complete genome sequences in the National Center for Biotechnology Information (NCBI) database were retrieved by blasted mcr genes (data as of October 2021). Due to low sequence identity between mcr genotypes and high sequence similarity between genotyped variants, a representative sequence for each genotype was selected for blasting, and all plasmid sequences hit were collected and incorporated into the alternative dataset before dereplication.
-
Plasmid Inc types and ARGs were identified by PlasmidFinder 2.1 (
https://cge.cbs.dtu.dk/services/PlasmidFinder/ ) (7) and ResFinder 4.1 (https://cge.cbs.dtu.dk/services/ResFinder/ ) (8). -
The coding sequences of plasmids were collected and the nonredundant homologous gene set was calculated by CD-HIT (9). Then, a matrix with rows and columns of the selected plasmid and nonredundant homologous genome was constructed. When a homologous gene was identified on the plasmid, 1 was input into the corresponding position in the matrix, coverage was set at 60% and the value was set at 0.00001. Otherwise, 0 was entered. Finally, the matrix was output as a gene clustering tree and displayed by iTOL v6 (https://itol.embl.de/) (10).
-
After manual alignment of the plasmid sequences, genome alignment and display were performed using the Mauve plugin (11) in Geneious Prime (v2021.2). The seed weight calculation and the minimum LCB score were set to automatic. The evolutionary tree was constructed using UPGMA for the tree building method and Tamura Nei for the genetic distance model.
-
The world map was obtained from the standard map service system of the Ministry of Natural Resources (
http://bzdt.ch.mnr.gov.cn/ ). -
The 455 pMCR complete genome sequences in this study were collected from 44 countries across 6 continents from 1998 to 2020 (Supplementary Figure S1A). The plasmid hosts involved 30 species of 15 genera, and Escherichia accounted for the vast majority (234, 51.4%), and about half of the plasmids were isolated from Escherichia coli (232, 51.0%) (Supplementary Figures S1B and S2).
In these plasmids, 52 incompatible types (Inc) were identified, of which 5.9% were unknown. IncHI2 (135, 29.7%), IncI2 (107, 23.5%), and IncX4 (66, 14.5%) were the 3 major Inc types and had wide national distribution (Supplementary Figures S1C and S3). Interestingly, 62 (13.6%) plasmids were fusion plasmids composed of at least 2 Inc types (Supplementary Figure S1C).
Eight mcr genotypes were found in this dataset. mcr-1-carrying plasmids (271, 59.6%) accounted for most of the collection (Supplementary Figure S1D, and S4). In this dataset, the mcr genotypes tended to correspond to some common plasmid Inc types. In IncI2 and IncX4 plasmids, only mcr-1 was found. IncHI2 had mcr-1 or mcr-9, and IncP1 carried mcr-1 or mcr-3 (Figure 1). The remaining Inc types did not show a high association with mcr genotypes due to the small sample size. Additionally, the host bacteria of IncX4 plasmids were narrow, with only E. coli, S. enterica, and K. pneumoniae, and the host range of IncHI2 was broad (Figure 1).
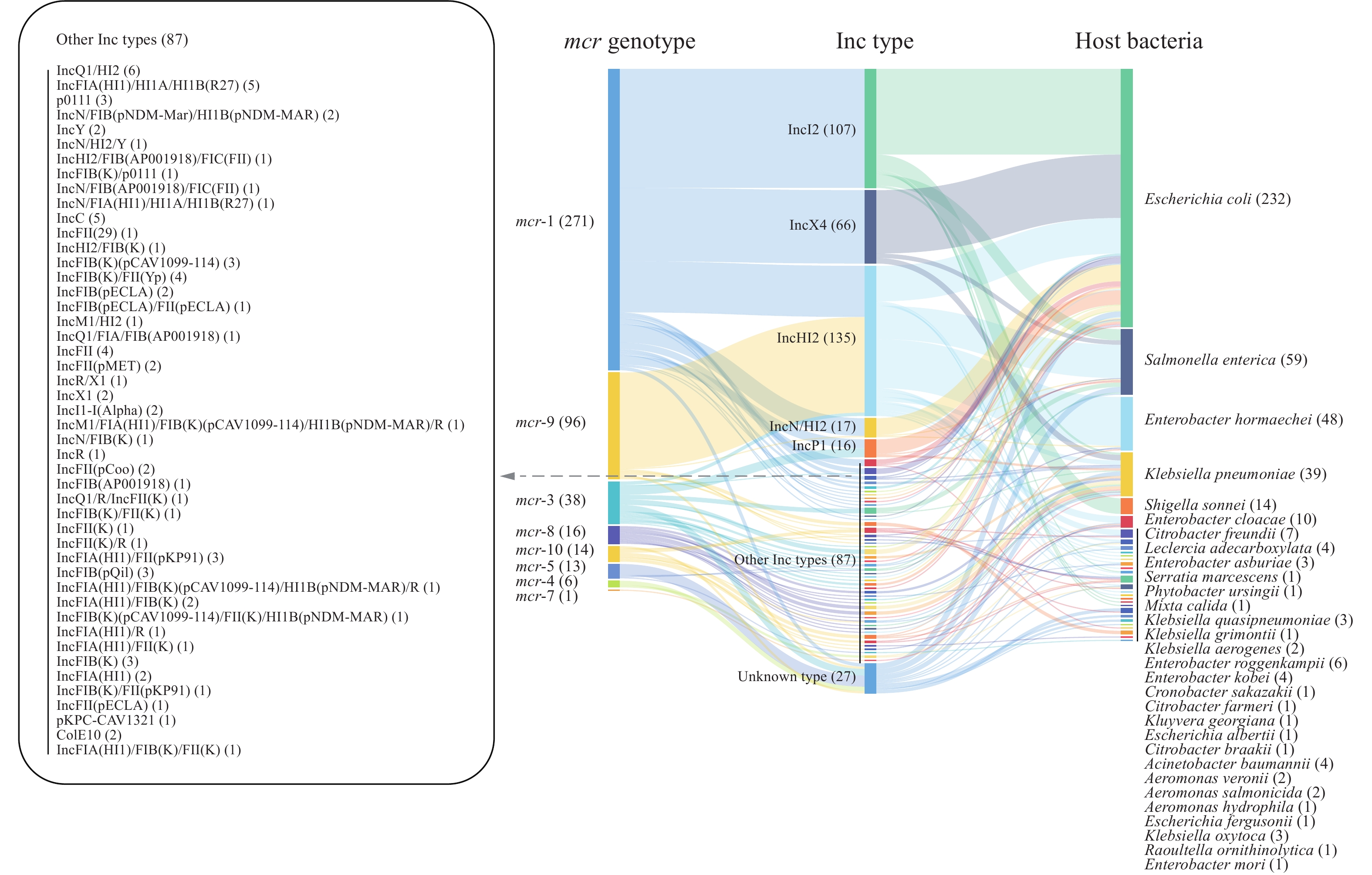 Figure 1.
Figure 1.The correspondence between mcr genotypes, Inc types, and hosts.
Note: The numbers in brackets represent the number of plasmids. Other Inc types are expanded in the left box. -
Except for mcr, a total of 97 ARGs carried by pMCRs were identified. The average number of ARGs was 3.9 per plasmid, and 48.4% of the plasmids carried two or more ARGs including mcr genes (Figure 2A). The average number of ARGs carried by IncHI2 and IncC pMCRs was 6.8 and 7.0 per plasmid, respectively. In contrast, IncI2, IncX4, and IncP1 carried low numbers of ARGs, approximately 1.0 to 1.2 per plasmid, respectively (Figure 2A). In addition, the number of ARGs carried by fusion plasmids was significantly higher than the single Inc plasmid (Figure 2B, P<0.0001).
 Figure 2.
Figure 2.The comparison of the amounts of ARGs carried by each Inc type of pMCR and the carrying rate of ARG classes. (A) The number of ARGs carried by each Inc type of pMCR. (B) The comparison of the amount of ARGs carried by single Inc type plasmids and multi-Inc type fusion plasmids. (C) The carrying rate of ARG classes.
Note: (A) The Inc type is arranged in descending order according to the median number of carrying ARGs. Black dots represent outliers. The number in parenthesis represents the amount of plasmids contained in each type. Abbreviations: ARGs=antimicrobial resistance genes; pMCRs=mcr-carrying plasmids.The ARGs found in these pMCRs were involved in the resistance to 14 classes of antibiotics. The ARGs related to folate pathway antagonist resistance genes (40.2%), β-lactam resistance genes (39.8%) and aminoglycoside resistance genes (37.1%) were the most common genes to cooccur with mcr genes in these plasmids (Figure 2C).
-
Genomes of the pMCRs in the dataset were compared to show genome similarities. Based on their gene components and sequences, a pangenome cluster tree was constructed by the BLAST matrix of the nonredundant coding gene set with the setting values (Figure 3). Inc typing showed a better association with clustering. In IncX4 and IncI2, pMCRs had relatively conservative genomes; in contrast, IncHI2 pMCRs presented much higher divergence. pMCRs isolated from different specific hosts were scattered in various clusters, and no host clustering was observed (Figure 3).
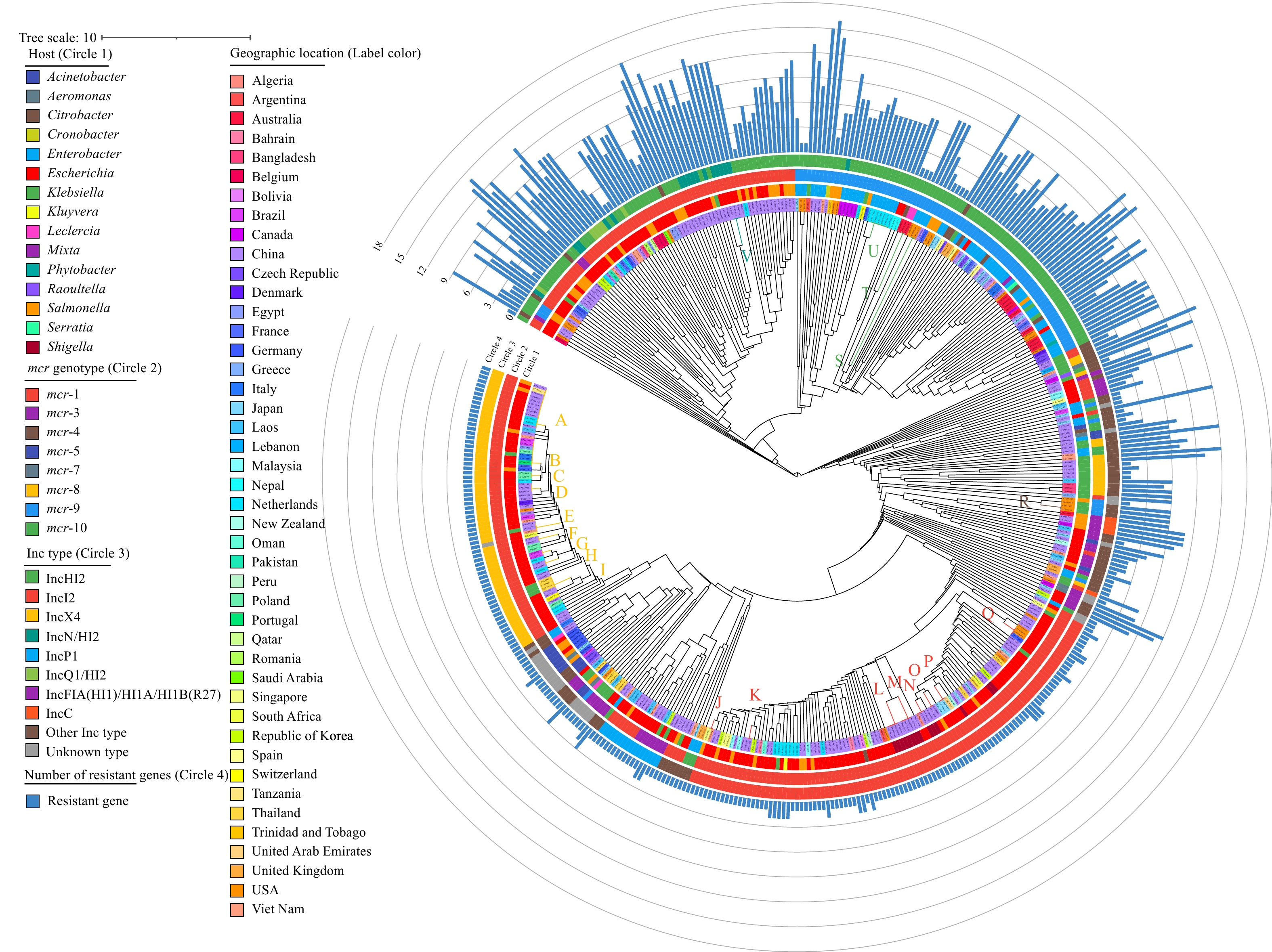 Figure 3.
Figure 3.The Pangenome tree of 455 pMCRs.
Note: Circles 1 to 4 represent hosts, mcr genotypes, plasmid Inc types and the amounts of ARGs, respectively. Countries of sources are marked on the sample codes. Abbreviations: ARGs=antimicrobial resistance genes; pMCRs=mcr-carrying plasmids.In the pangenome cluster tree, some pMCRs with high similarities in gene content and sequences were grouped into 22 clusters (named Cluster A to V) (Supplementary Figure S5). Then, the genome alignment among the plasmid in each cluster revealed the potential spatiotemporal transmission and cross-host transfer of pMCRs. The main differences between pMCR genomes in most clusters were the single nucleotide polymorphisms and fragment indels in some clusters, suggesting their very close evolutionary relationship and even possible epidemiological association (Supplementary Figure S5). In these clusters, Cluster A, contained 12 IncX4 pMCRs with basically the same length, and only a few SNPs were identified among the plasmids (Figure 4A). The plasmids were isolated in three countries (China, Tanzania, and the Netherlands), various sources (patients, food, livestock, and the environment), and different host bacteria (E. coli and S. enteritidis), which implied that there might be a trans-spatiotemporal resistance epidemic event initiated by the same plasmid clone (Figure 4). Similar transmission events were also observed in Clusters B, C, E, and others (Figure 4) In some IncI2 plasmid clusters, such as Clusters J and K, inversions of gene segments occurred between plasmids (Figure 4A). In Cluster J, inversions of gene segments were identified by the plasmid genome alignment, although the plasmids only differed in length by 7 bp, and the same recombination position was also observed between pMCRs in Cluster K (Supplementary Figure S6). IncHI2 plasmids accounted for the largest proportion in the complete genome dataset of pMCRs, and 3 highly conserved clusters (Cluster S, T and U, Supplementary Figure S5) were identified. A total of 6 isolates of human origin pMCRs isolated from the Netherlands were included in Cluster U, with a less than 3 bp difference in length (Figure 4A). The genome alignment showed that plasmid CP071022.1 had a 161 kbp fragment inversion compared to the others. This study did not find the origin and reservoir of these plasmids, but their sources from different hospitals and years strongly suggested the spread and risk of MDR mediated by these plasmids (12). The combination of pangenome clustering and genome alignment in this study revealed the accumulation of mutations and the recombination of genome fragments in the epidemic process of resistant plasmids.
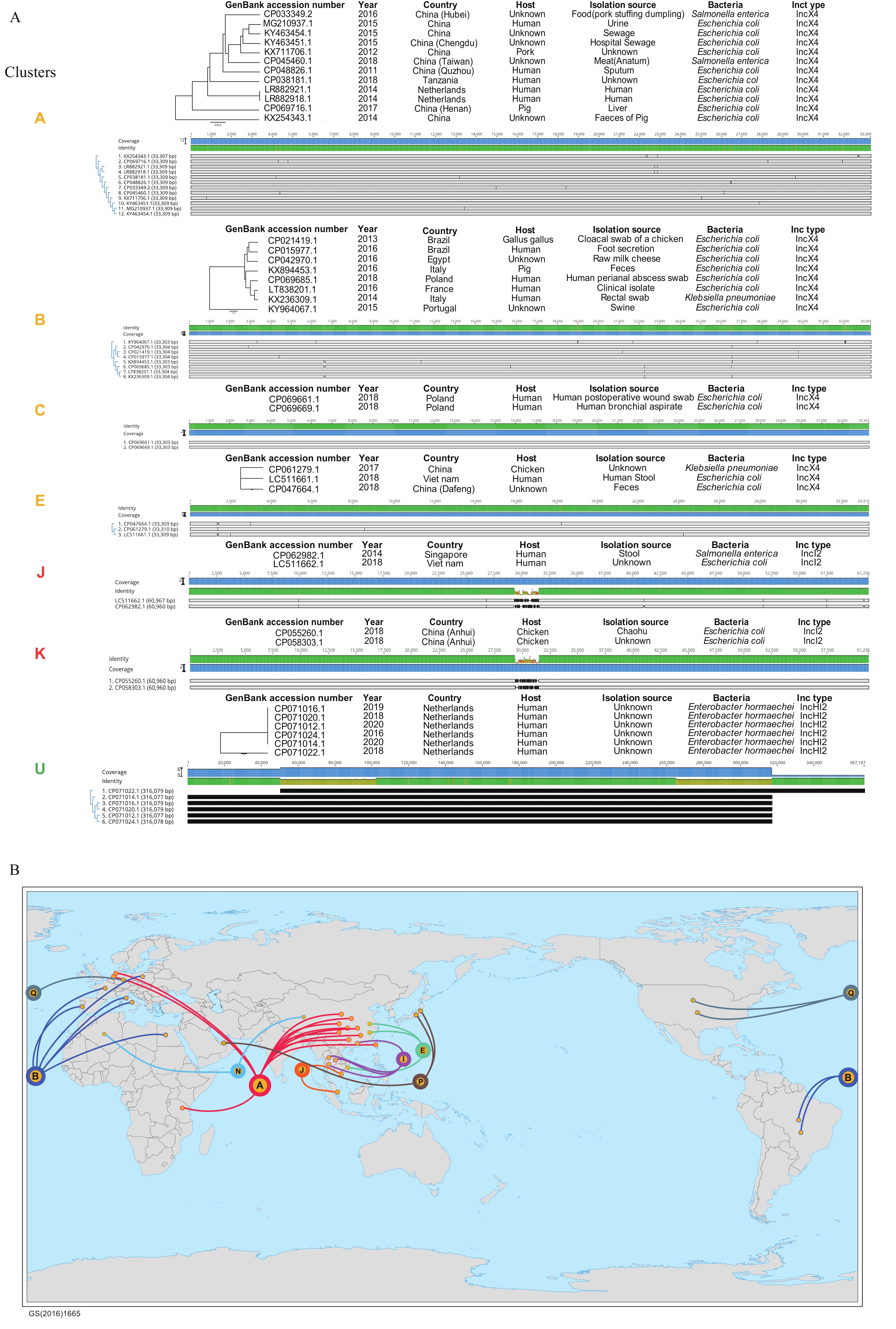 Figure 4.
Figure 4.Representative plasmid clusters among 22 clusters with high complete plasmid sequence similarity and the geographical distribution of transnational epidemic clusters. (A) Representative plasmid clusters among 22 clusters with high sequence similarity. (B) The geographical distribution of transnational epidemic clusters.
Note: (A) The data of each cluster consists of pMCR isolation information (isolation year, country, host, sample source, bacterial hosts, and Inc types) and genome alignment results. (B) The area and color of the solid circle marked with letter represent the amount and Inc type of pMCRs in the cluster, respectively. Abbreviation: pMCRs=mcr-carrying plasmids. -
In Enterobacteriaceae, colistin resistance mediated by the plasmid-borne mcr genes spread rapidly in recent years (4,13). pMCRs are independent genetic elements that can transfer across hosts and have genomic plasticity, and complete sequences of pMCRs are required in the surveillance of the colistin resistant bacteria.
The complete plasmid sequence can definitely reveal the co-transfer of multiple ARGs on the same plasmid. In this study, half of pMCRs carried multiple ARGs, but some Inc type plasmids only carried mcr, such as IncX4. Association studies of plasmid Inc types with the ability to carry ARGs can be applied to evaluate the risk of multi-resistance for different Inc type resistance plasmids. In addition, complete plasmid sequences can clearly define the genes related to environmental adaptation and conjugation, such as heavy metal resistance genes and type IV secretion systems, which can be used to evaluate the maintenance and transfer ability of the resistant plasmid.
In epidemiological surveillance of resistance, it is necessary to identify the transmission events involving different countries, dates, sources, and hosts. Applying the pangenome clustering based on complete plasmid genome to the surveillance can help to associate independent data into transmission events. Even in nosocomial infection control, this monitoring mode can be used to reveal the source and spread of infection (14). Although the amount of pMCRs with complete genome sequences in the database is limited, we still observed some plasmid genome clusters with only some SNP differences among the 455 pMCRs by genome alignment. Among these clusters, some plasmids existed in a variety of host bacteria isolated from different countries, years, and sample sources, which provides evidence for epidemiological surveillance and tracing. These plasmids, which spread widely and involve various sources, have higher public health risks and should be monitored.
This study was subject to some limitations. Resistant bacteria carrying mcr genes have been identified worldwide. For the needs of plasmid type identification and gene composition analysis, the data set of this study only collected pMCR sequences with the complete genome from the NCBI database, which may lead to the deviation of plasmid characteristic statistics caused by sampling biases. Although the transmission of many resistant plasmids remained unobserved, we still found some representative pMCR transmission across countries and hosts. With the establishment and continuous improvement of the resistant plasmid surveillance network, more high-quality complete plasmid genomes will be provided for analysis.
The genome alignment based on complete plasmid genomes of pMCRs has revealed the epidemic events across countries, years, sources, and hosts, which suggests a potential dissemination of pMCRs among human, food, animal, and environment. Therefore, laboratory surveillance networks based on the genome sequencing of resistant plasmids are needed to monitor the epidemic and transmission of antimicrobial resistance. Methods of genome sequencing, assembly, and analysis strategies for resistant plasmids, such as typing and genome alignment, should be optimized and standardized to promote effective data collection, sharing, analysis, and application among network laboratories.
HTML
Collection of pMCRs with Complete Genome Sequences
Identification of Plasmid Inc Types and ARGs
Pangenome Tree
Plasmid Whole Genome Alignment
World Map
Diversification of Plasmid Inc Types and Wide Host and Geography Distribution of pMCRs
The High Cooccurrence Rates of ARGs in pMCRs
Plasmid Genome Clustering Showed Evidence of Spatiotemporal Transmission and Cross-Host Transfer of pMCRs
| Citation: |

|