
 Download:
Download:




HTML
-
Introduction: Most current research on Salmonella has targeted prevalent serotypes, such as S. Typhimurium and S. Enteritidis, but the epidemiology and molecular characteristics of less prevalent serotypes remain insufficiently characterized. This study focused on S. Give, a less common serotype, to elucidate its genomic characteristics and antimicrobial resistance gene (ARG) profiles in China.
Methods: The whole-genome sequences of 185 isolates of S. Give were extracted from the Chinese Pathogen Identification Network database from 2017 to 2024 and subjected to ARG detection and phylogenetic analysis.
Results: Two major sequence types (STs) were identified among the S. Give isolates, with ST516 being the predominant ST (92.43%) in China — consistent with the global ST distribution, except in the U.S., where ST654 prevailed (82.70%). The multidrug resistance (concurrent carriage of ≥3 ARGs) rate was 3.51%. All 185 isolates harbored the T57S point mutation in the parC gene on the chromosome, and an increasing trend was observed in the quinolone resistance gene qnrB19 prevalence in China from 2020 to 2024. In the major sublineage, 80% of the isolates contained the qnrB19 gene, and 86.41% of the isolates carried the small mobilizable plasmid Col (pHAD28) harboring the qnrB19 gene. Six clusters were detected, indicating several potential outbreaks within China. Moreover a close phylogenetic relationship with European strains was exhibited.
Conclusion: This study shows that S. Give predominates in China and is characterized by clonal expansion and the widespread presence of qnrB19-harboring plasmids. S. Give’s sporadic outbreaks and multidrug resistance represent emerging public health threats. Moreover, the ongoing genomic surveillance of uncommon serotypes is essential to identify and mitigate concealed risks to public health.
-
Salmonella spp. infections remain a major foodborne disease with global economic and health burdens (1–3). Current studies have predominantly targeted the epidemiology and molecular characteristics of prevalent serotypes, such as Salmonella Typhimurium, Salmonella Enteritidis, and Salmonella 4,[5],12:i:- (4–5), leaving gaps in the understanding of less prevalent serotypes. Salmonella Give, one of the rare serotypes of Salmonella, has caused hundreds of cases annually in Europe in recent years (the reported incidence is 0.02/100,000 to 0.08/100,000 from 2007 to 2024, https://atlas.ecdc.europa.eu/public/index. html). In the United States, S. Give is the 38th most prevalent serotype among Salmonella isolates in humans. Although S. Give is not the dominant serotype globally (6), its sporadic occurrence and localized outbreaks indicate its potential public health significance (7–8), warranting further investigation. S. Give ranks 14th among all serovars in China, with isolates recovered from both human and non-human sources (9); however, the prevalence of antimicrobial resistance (AMR), genetic characteristics, and transmission dynamics have rarely been reported. This study aimed to describe the genomic epidemiology and antimicrobial determinants of S. Give to provide a basic understanding of this serovar. This study used whole-genome-based approaches on national and global genomes to describe the genotypes, population structures, and AMR genes of S. Give. This study identified clonal expansion with the co-occurrence of an increasing prevalence of qnrB19 and potential outbreaks in China.
-
From 2017 to 2024, 15,731 Salmonella genomes were isolated and recorded in the National Pathogen Identification Network (CPIN) database. The whole-genome sequences and geographical data of 185 S. Give strains with geographical data, ranked 14th in the database. Most isolates (173/185) were recovered from the feces of patients with diarrhea, 10 from food sources, and 2 from unknown sources (
Supplementary Figure S1 , availablehttps://weekly.chinacdc.cn/ ). -
Multilocus sequence typing (MLST) was performed using PubMLST (https://pubmlst.org/). The ARGs were determined by ResFinder 4.1.0 (https://genepi.food.dtu.dk/resfinder) with the screening criteria set as sequence identity ≥80% and coverage ≥60% to ensure the reliability of ARG annotations. Multidrug resistance (MDR) was defined as the concurrent carriage of ≥3 ARGs that individually belonged to different categories of antimicrobial agents. Plasmids were detected and compared via PlasmidFinder 2.1.0 (http://cge.cbs.dtu.dk/services/PlasmidFinder/) and NCBI BLASTn, respectively, and the resulting plasmids (≥95% identity, ≥90% coverage) were visualized linearly using Easyfig (https://mjsull.github.io/Easyfig/)
-
Quality control was conducted using Quast 5.0.2 (10) after retrieving the assembled genomic sequences from CPIN and EnteroBase, resulting in two genome subsets of S. Give: 185 domestic and 1,446 global genomes (https://enterobase.warwick.ac.uk/). Single-nucleotide polymorphisms (SNPs) in the core genome were determined with Snippy 4.4.5 (https://github.com/tseemann/snippy). The reference genome used in the phylogenetic tree was S. Give strain NCTC5778 (GCA_900477925.1), with a total length of 4,638,063 bp. SNP calling was performed using Snippy 4.4.5 (https://github.com/tseemann/snippy) with default parameters, resulting in about 7,000 core SNPs.
A maximum likelihood tree based on core-genome SNPs was constructed after removing recombinants using the Gubbins software 3.3.5 (https://github.com/nickjcroucher/gubbins) (11) and was presented using iTOL (https://itol.embl.de). Cluster analysis of plasmids carried by the strains was performed using the Average Nucleotide Identity (ANI) 1.34.
-
MLST of S. Give revealed that the sequence type ST516 was predominant (92.43%), followed by ST654 (5.9%), and three novel sequence types were identified in China (
Supplementary Figure S1 ). Human-derived isolates constituted the majority of both ST516 (94.74%) and ST654 (90.91%) cases, whereas isolates from food sources were relatively rare (4.09% for ST516 and 9.09% for ST654;Supplementary Figure S1 ). ST516 was distributed across 24 provincial-level administrative divisions (PLADs), with a slightly higher prevalence in the eastern and southern regions than in the other regions. Specifically, the prevalence of ST516 in eastern China was 95.93%, which was higher than that in central (84.62%) and western (86.96%) China, with a prevalence of 94.26% exceeding that in northern China (88.89%). In contrast, ST654 was more frequently detected in central (12.5%) and western (13.04%) regions than in eastern China (2.46%; Figure 1A). Temporally, ST516 was first detected in eastern China and was subsequently identified in the central and western regions in this study (Figure 1A). ST654 was initially reported in 2021 (eastern region) with subsequent detections in 2023 (central and western regions) and 2024 (central region), demonstrating a progressive increase in the central region and a transient increase followed by a decline in the eastern and central regions (Figure 1A).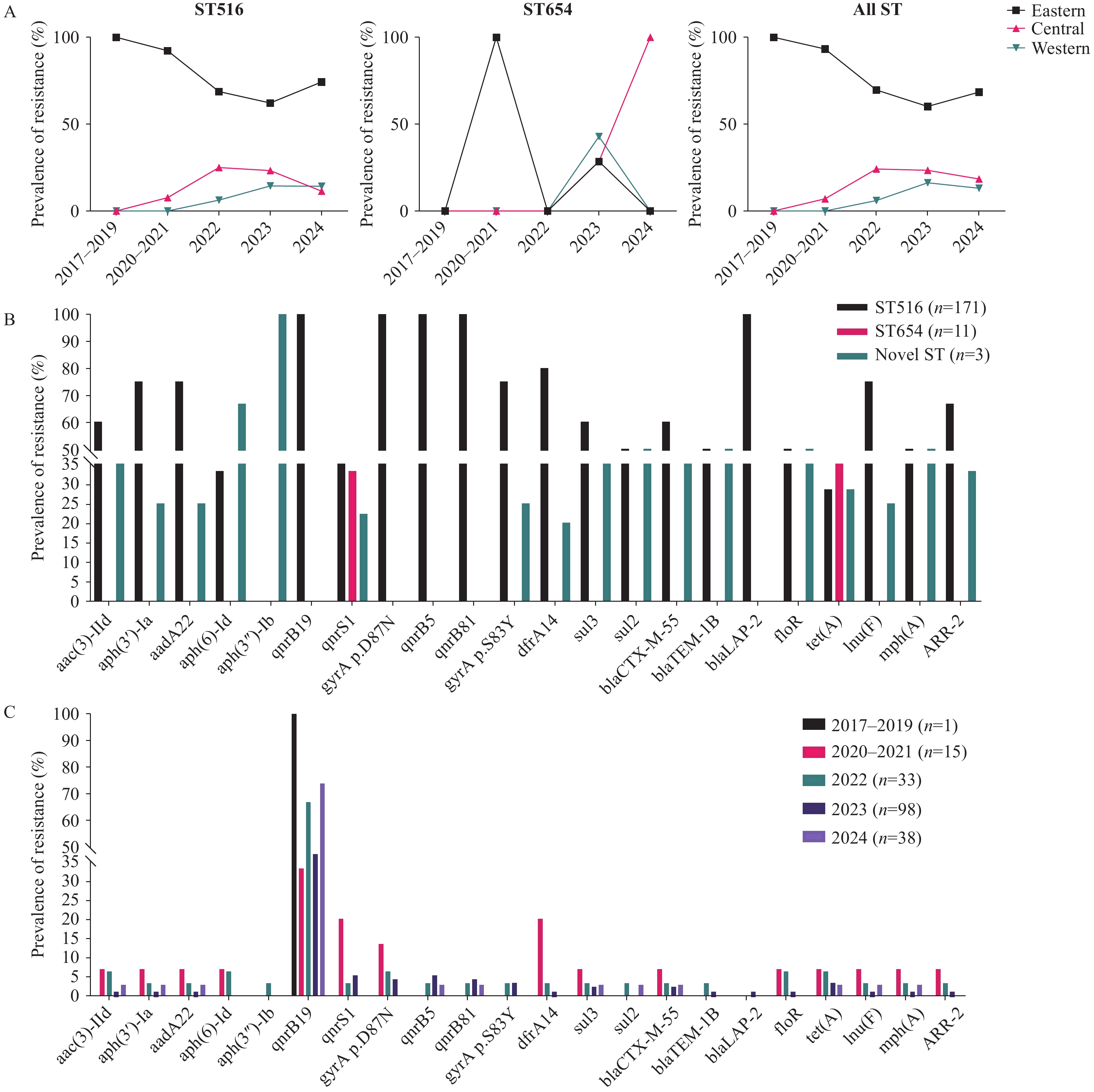 Figure 1.
Figure 1.Dynamic distribution of STs and their associated ARGs. (A) shows the temporal trends of ST516, ST654, and all strains across the eastern, central, and western regions. (B) depicts the proportions of different resistance genes within various STs. (C) illustrates the proportions of different resistance genes throughout the years.
Abbreviation: ST=sequence type; ARG=antimicrobial resistance gene. -
Analysis of 185 S. Give isolates revealed 24 ARGs across nine categories. All isolates harbored the aminoglycoside resistance gene aac(6’)-Iaa and the fluoroquinolone resistance-associated mutation parC p.T57S. Approximately 55.68% of the isolates carried the quinolone resistance gene, qnrB19, whereas other resistance genes were infrequent (<5%;
Supplementary Table S1 ). ST516 exhibited the broadest ARG repertoire, with 14 ARGs present in >50% of the isolates (Figure 1B). In contrast, ST654 displayed limited diversity and prevalence of resistance genes, except for the universal presence of parC p.T57S and aac(6’)-Iaa (both 100%; Figure 1B). The MDR rate was 3.51%, which was primarily associated with the presence of parC p.T57S, dfrA14, floR, and sul3 and was exclusively observed in ST516. The extended-spectrum β-lactamase (ESBL) gene blaCTX-M-55 was detected in five isolates from five PLADs. These isolates also harbored sul3, qnrS1, aac(3)-IId, lnu(F), ARR-2, and tet(A), which confer resistance to third-generation cephalosporins, sulfonamides, fluoroquinolones, aminoglycosides, lincosamides, and tetracyclines. Although the number of isolates varied across the years, a noticeable increase in qnrB19 detection was observed in the later years of surveillance, whereas many other ARGs appeared stable or fluctuated at low levels (Figure 1C). -
A phylogenetic tree based on core-genome SNPs revealed that the 185 S. Give strains formed two lineages: A (11 strains) and B (174 strains). All isolates in lineage A were classified as ST654, whereas lineage B comprised ST516 and three novel sequence types. Lineage B was further subdivided into three sub-lineages (Figure 2). Sublineage 1 included 35 strains distributed across 12 PLADs in three regions over five years and was characterized by a broad spectrum of resistance genes up to 13 types; (
Table 1) , although the prevalence of each gene was low (2.86%–22.86%). Sublineage 2 (10 strains) was restricted to two PLADs in the eastern region over three years, with no resistance genes detected. Sublineage 3 represents the predominant clonal group (129/174; 74.14%), spanning all 24 PLADs from 2017 to 2024. This sublineage also harbored multiple resistance genes up to 13 types; (Table 1 ), with a notably higher carriage rate of qnrB19 (79.84%) than that of other genes (0.78%–5.43%). Furthermore, 86.41% of qnrB19-positive strains in sublineage 3 carried the Col (pHAD28) plasmid (2.6kb) encoding qnrB19 (Figure 2). In contrast, lineage A (ST654) contained only tet(A) and qnrS1 (excluding the parC mutation and aac(6’)-Iaa), each with a carriage rate of 27.27%. Temporal and geographic distributions of the three sublineages varied across PLADs and years. For example, in Shandong and Sichuan provinces, sublineage 1 predominated in 2021, but was supplanted by sublineage 3 in 2022 and 2023, respectively.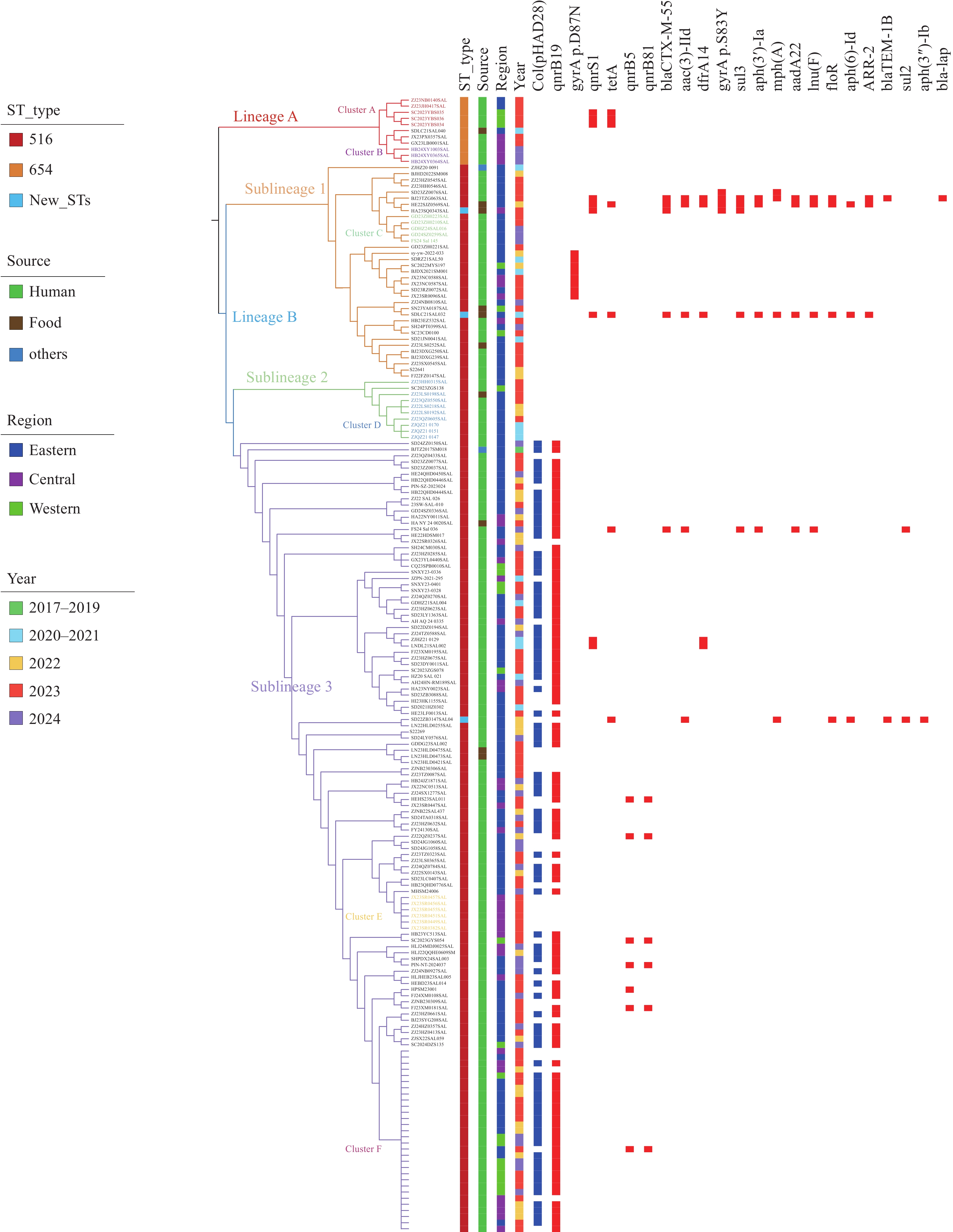 Figure 2.
Figure 2.Phylogenetic tree of 185 S. Give isolates in China from 2017 to 2024 based on core-genome SNPs.
Note: The evolutionary tree is divided into two lineages: lineage A (red) and lineage B. Lineage B is further subdivided into three parts: sublineage 1 (yellow), sublineage 2 (blue), and sublineage 3 (purple).Six clusters coexist on the evolutionary tree: Cluster A (red), Cluster B (purple), Cluster C (green), Cluster D (blue), Cluster E (yellow), and Cluster F (blue-purple). The evolutionary tree is organized from the left to right layers as follows: STs (ST516, ST654, and three new STs), source (human, food and others), region (eastern, central, and western), year (2017-2019, 2020-2021, 2022, 2023, and 2024), and plasmid (Col(pHAD28)) and resistance genes (in order: qnrB19, gyrA p.D87N, qnrS1, tetA, qnrB5, qnrB81, blaCTX-M-55, aac(3)-IId, dfrA14, gyrA p.S83Y, sul3, aph(3')-Ia, mph(A), aadA22, lnu(F), floR, aph(6)-Id, ARR-2, blaTEM-1B, sul2, aph(3'')-Ib, and bla-LAP2).
Abbreviation: SNPs=single-nucleotide polymorphisms.
lineage ARGs profile Number of ARGs Number of Strains Lineage A qnrS1-tet(A) 2 3 gyrA p.S83Y 1 1 No ARGs − 6 Lineage B Sublineage 1 gyrA p.D87N 1 6 gyrA p.S83Y 1 3 mph(A)-gyrA p.S83Y 1 1 qnrS1-sul3-blaCTX-M-55 2 1 blaTEM-1B-floR-qnrS1-sul3-aac(3)-IId-mph(A)-dfrA14-
aph(3')-Ia-blaCTX-M-55-ARR-2-aadA22-blaLAP-2-lnu(F)3 1 floR-qnrS1-sul3-aac(3)-IId-tet(A)-aph(6)-Id-dfrA14-
aph(3')-Ia-blaCTX-M-55-ARR-2-aadA22-lnu(F)13 1 floR-qnrS1-sul3-aac(3)-IId-tet(A)-mph(A)-aph(6)-Id-
dfrA14-aph(3')-Ia-blaCTX-M-55-ARR-2-aadA22-lnu(F)12 1 No ARGs − 22 Sublineage 2 No ARGs − 10 Sublineage 3 qnrB19 1 90 qnrB19-qnrB81-qnrB5 3 6 qnrB19-qnrS1-dfrA14 3 2 gyrA p.D87N 1 2 qnrB19-gyrA p.D87N 2 2 qnrB19-sul2-sul3-aac(3)-IId-tet(A)
-aph(3')-Ia-blaCTX-M-55-aadA22-lnu(F)9 1 qnrB19-gyrA p.S83Y 2 1 qnrB19-qnrB5 2 1 blaTEM-1B-floR-sul2-aac(3)-IId
-tet(A)-mph(A)-aph(6)-Id-aph(3'')-Ib8 1 No ARGs − 23 Table 1. The situation of antibiotic resistance gene carriage in different branches of the phylogenetic tree of 185 strains.
Six clusters of S. Give were detected in China. Two clusters, A and B, were located within lineage A. Cluster A comprised five ST654 strains differing by 2–3 SNPs (Figure 2), which were isolated from three cities (Yibin, Ningbo, and Jinhua) in May 2023, suggesting an interprovincial outbreak. Cluster B consisted of three closely related strains (0–3 SNP differences) detected in Xiangyang, Hubei Province, in 2024, which was consistent with a localized outbreak. Cluster C, within sublineage 1, comprised five strains differing by <5 SNPs and was identified in Guangdong Province. Cluster D, comprising nine strains with 0–5 SNPs differences, was found in Zhejiang Province, indicating another regional outbreak. Within sublineage 3, cluster E consisted of six qnrB19-negative strains (0–5 SNP differences) collected in Jiangxi Province (Figure 2), which were phylogenetically adjacent to qnrB19-positive strains and unique among the clusters, with all others carrying qnrB19. Additionally, Cluster F, comprising 30 closely related isolates differing by only 0–2 SNPs, was identified in the same sublineage (Figure 2).
ANI clustering analysis was performed on the Col(pHAD28) plasmids harboring 89 strains, grouping them into six clusters (A–F) (https://enterobase.warwick.ac.uk/). The intracluster similarity ranged from 97.29% to 100.00%. The sequence of the Col(pHAD28) plasmids (2,699 bp+) displayed high identity with 600 plasmids from 10 genera and 15 species. This indicates that small ColE1/Col440I-like plasmids (2.6–3.0 kb), carrying almost exclusively the qnrB19 gene, are highly conserved and widely distributed across diverse bacterial species (12–14).
-
Among the 1,631 global isolates of S. Give, five major STs were identified, of which ST516 was the most prevalent (45.62%), followed by ST654 (42.49%). ST654 was almost exclusively concentrated in the United States (82.70%), whereas ST516 was the predominant ST in all other countries, including China (Figure 3, and https://enterobase.warwick.ac.uk/). Additionally, ST2589, ST524, and ST2227 exhibited distinct regional distribution patterns; for example, ST2589 was mainly detected in Mexico (22 isolates, 22%), whereas ST524 was largely confined to the UK (27 strains, 34.2%).
 Figure 3.
Figure 3.Phylogenetic tree of 1,631 global S. Give isolates from 1935 to 2024 based on core-genome SNPs.
Note: The evolutionary tree constructed on the basis of core genome multilocus sequence analysis is organized from the inner to outer layers as follows: Seven continents/regions (China, North America, Europe, Asia, Africa, South America, and Oceania); Sequence types (STs): Major STs including ST516, ST654, ST524, ST2589, ST2227, and other STs; Temporal distribution: Five time periods (1931–2000, 2001–2005, 2006–2010, 2011–2015, and 2016–2024); Source: Five categories (Human, Environment, Food, Animal, and Poultry). The phylogenetic tree is primarily divided into three lineages: lineage 1 (415 strains) with a blue background color, lineage 2 (456 strains) with a yellow background color, and lineage 3 (760 strains) with a green background color. Chinese strains are labeled in red and clustered within lineage 1 (139/185).
Abbreviation: SNP=single-nucleotide polymorphism; ST=sequence type.
The phylogenetic analyses revealed three global lineages (Figure 3). Lineage 1, dominated by ST516 (95.90%), is mainly distributed in Europe and Asia, with human-derived isolates being the primary source, followed by animal and food sources. Lineage 2 consisted mostly of ST516 (75.89%) and ST524 (17.32%), which exhibited a broad geographical range and balanced representation across different sources, with animal isolates being the most common. Lineage 3 was primarily composed of ST654 (91.18%), which is concentrated in North America, and over 70% of its isolates were from environmental sources. Chinese isolates were predominantly clustered in lineage 1, with only a scattered distribution in lineages 2 and 3. The phylogenetic reconstruction of 744 ST516 strains indicated a close genetic relationship between the Chinese isolates and UK food-derived strains (
Supplementary Figure S3 ), suggesting a shared common ancestor and potential transnational transmission via the food chain.Similar to the domestic strains, all global isolates carried the aac(6’)-Iaa gene and parC p.T57S mutation, whereas only 8.95% of the isolates harbored the qnrB19 gene. Among the qnrB19-positive isolates, most (70.55%) originated in China, followed by Nigeria (18%), France (6.87%), and the UK (6.25%). These isolates were predominantly assigned to ST516 and clustered in lineages 1 and 2, but were nearly absent in lineage 3 (Figure 3). Additionally, the source of qnrB19 varied geographically. In China and Nigeria, qnrB19 was almost exclusively detected in human isolates (100% of qnrB19-positive isolates from both countries were human-derived), whereas in the UK and France, qnrB19 was detected more frequently in animal than human-derived isolates (Figure 3). Specifically, the detection rate of qnrB19 in animal-derived isolates was 55.56% in the UK and 63.64% in France, compared with 27.78% and 36.36% in human-derived isolates from the UK and France, respectively (Figure 3). Notably, 95.21% of qnrB19-positive strains were recovered during the past decade, indicating a recent global increase in the spread of qnrB19-carrying S. Give. Furthermore, 129 of 146 qnrB19 alleles were located on the Col(pHAD28) plasmid, of which 89 (68.99%) were detected in China (Figure 2, Figure 3 and
Supplementary Table S1 ), suggesting the ongoing domestic circulation of this small mobilizable resistance plasmid in S. Give. Notably, among the global genomes, 22 environmental isolates (30.14% of all environmental samples) and 19 food isolates (26.03% of all food samples) harbored multiple resistance genes together with the chromosomal gyrA p.S83Y mutation (Figure 3, https://enterobase.warwick.ac.uk/), suggesting that MDR-associated determinants were present in both environmental- and food-associated S. Give populations. -
This study provided a comprehensive analysis of the phylogenetic structure, spatiotemporal dissemination, and AMR profiles of S. Give in China. These findings reveal the high global genetic diversity of S. Give and identify the presence and increasing prevalence of an ST516 clone in China that is associated with the mobile fluoroquinolone resistance gene, qnrB19.
The majority of S. Give isolates in this study were pan-susceptible to antibiotics; however, there were several MDR isolates of ST516 in China. This genotype is characterized by various resistance genes, including those associated with chloramphenicol (floR), sulfonamide (sul3), cephalosporin (blaCTX-M-55), and quinolone resistance (qnrB19, qnrS).
qnrB19 is widely distributed across diverse enterobacteria and is typically located on small plasmids such as IncQ, IncC, and Col (15). In the United States, approximately 95.4% of waterborne S. Enteritidis isolates carry the plasmid-mediated qnrB19 (16). Similarly, in Poland, qnrB19 has been identified as a key quinolone resistance gene that requires monitoring across multiple Salmonella serotypes (17). The plasmid Col(pHAD28) carrying qnrB19 has been detected in S. Kentucky from poultry in Nigeria, including in environmental and clinical isolates from Ukraine and Bangkok (18–20), indicating its potential for dissemination across geographic regions and host species. In contrast to the predominant serotypes in China (Salmonella 4,[5],12:i:-), the prevalence of qnrB19 is <1%, with qnrS1 being more frequently detected (4). Studies have shown that qnrB19 alone can increase the minimum inhibitory concentration (MIC) of ciprofloxacin 10-fold and moxifloxacin 8-fold in vitro (21). When combined with the gyrA mutation S83F/D87N, qnrB19 synergistically enhances fluoroquinolone resistance by 2-2.5 fold (22). However, the interaction between qnrB19 and parC p.T57S remains unclear and requires further investigation.
Col(pHAD28) is non-self-transmissible (23), and its mobilization depends on conjugative plasmids such as IncHI2 or IncF, which provide a type IV secretion system (T4SS) for horizontal transfer, which may explain the relatively low qnrB19 prevalence in dominant Chinese Salmonella serotypes. However, recent surveillance data have shown an upward trend in qnrB19 carriage among S. Typhimurium and Salmonella 4,[5],12:i:- in China, suggesting a potential increase in quinolone resistance mediated by this gene. A global genomic analysis of 47,452 Salmonella isolates (1905–2020) revealed a low overall prevalence of qnrB19: 0.43% for qnrB19 alone and 0.87% for qnrB19-parC p.T57S combination (24). A review of 22 publications involving 468 strains revealed the widespread distribution of qnrB19 across 49 Salmonella serotypes in nine countries ( 339 strains from the Americas, 63 from Europe, and 67 from Africa, 25-28). These findings are consistent with the data on qnrB19 in Asia (excluding China), where only 33 human-derived strains have been identified (primarily from Brazil and Nigeria). Except for one strain lacking the ColE-like replicon, all qnrB19-positive strains carried Col(pHAD28), indicating a strong association between qnrB19 and this plasmid type in Salmonella.
Epidemiological data have highlighted the public health significance of S. Give in China. S. Give accounts for 1.18% of human-derived Salmonella isolates (ranking 13th); based on the national salmonellosis incidence of 1,295.59/100,000 (29), the estimated burden of S. Give is approximately 15/100,000, substantially higher than the incidence of typhoid and paratyphoid fevers (≈0.5/100,000). Additionally, S. Give genomes constitute 0.17%–0.27% of the global Salmonella genomic database submissions (as of June 17, 2023), suggesting that its incidence or disease burden in China exceeds the global average and is higher than that in developed regions such as Europe and the United States. Combined with the emergence and clonal expansion of genotype ST516 linked to the increasing prevalence of qnrB19, this highlights public health threats.
This study had several limitations. Chinese strains were primarily derived from human sources, which may have introduced a selection bias. Second, the lack of clinical metadata (e.g., disease outcomes, exposure history, and travel history) limits insights into the factors influencing resistance patterns and transmission.
In conclusion, to our knowledge, this is the first genetic analysis of S. Give. This study reveals important information about serovar AMR and population structure. The results revealed that the number of ARGs harbored by S. Give was relatively limited; however, qnrB19 prevalence was high among both domestic and international isolates. These findings underscore the necessity for continuous and robust genomic surveillance to detect and mitigate concealed risks to public health.
-
We gratefully acknowledge all staff from the relevant provinces for their participation in Salmonella isolation.
-
This study does not involve human subjects, animals, or any clinical research. Therefore, ethical approval from an ethics committee is not required.
Data Source
Genotyping and Antimicrobial Resistance Gene (ARG) Detection
Phylogenetic Analysis
Sequence Type Distribution and Geographic Spread of S. Give in China
Quinolone Resistance and ARG Profiles of S. Give in China
Clonal Expansion and Widespread Carriage of the qnrB19 Gene, Especially Plasmid-mediated qnrB19 Dissemination, in China
S. Give Exhibited Geographical and Source Clustering Worldwide, with a Potential Global Increase in qnrB19-harboring
| Citation: |

|