
 Download:
Download:




-
Human adenoviruses (HAdVs) are non-enveloped, double-stranded DNA viruses with a genome size of approximately 35–36 kb (1). To date, 117 types have been identified and classified into seven species (A–G; http://hadvwg.gmu.edu/). Due to their diverse tissue tropisms, HAdV infections are associated with various illnesses, including acute respiratory infections, gastroenteritis, and conjunctivitis (2). Respiratory infections are frequently associated with specific species, including B (HAdV-3, 7, 14, 21, and 55), C (HAdV-1, 2, 5, and 6), and E (HAdV-4) (3).
HAdV type 14 (HAdV-14) was first identified in 1955 in a military recruit with acute respiratory disease (ARD) in the Netherlands, with only limited reports documented over subsequent decades (4). Approximately 50 years later, a novel and highly transmissible variant, designated human adenovirus type 14 variant p1 (HAdV-14p1), emerged and triggered multiple outbreaks in several countries, including the United States, the United Kingdom, Ireland, Canada, and Japan (5–9). These outbreaks affected both military and civilian populations and were associated with severe and fatal ARD cases (10). In China, HAdV-14p1 was initially detected in Guangzhou in 2010, with subsequent outbreaks in Beijing (2012), Liaoning (2012 and 2016), Gansu (2013), and Jiangsu (2015), resulting in substantial hospitalizations (11–15). HAdV-14p1 was sporadically detected in other provinces between 2011 and 2019. However, since 2019, no further cases have been reported in China.
Following the COVID-19 pandemic, HAdV-14 was newly identified in 2024 through sentinel surveillance in a pediatric patient hospitalized with bronchopneumonia in Chongqing, China. To elucidate the genetic characteristics of this re-emerging HAdV-14 strain and determine its phylogenetic relationship with previously circulating domestic and international strains, we sequenced its complete genome; and performed comprehensive phylogenetic and genetic variation analyses using publicly available global HAdV-14 genome data from GenBank.
-
Sentinel surveillance for acute respiratory infections conducted since 2019 and covering 15 provinces did not detect HAdV-14 until 2024. In 2024, the HAdV-14 strain Chongqing2024-115 was isolated from a sputum sample collected from a 4-year-old male patient hospitalized with acute bronchitis in Chongqing, China. The patient presented with primary symptoms of fever, cough, and expectoration and required a 10-day hospitalization. Chest imaging revealed no significant abnormalities in either lung. Initial pathogen screening was positive for HAdV, and subsequent sequencing and analysis of the penton base, hexon, and fiber genes confirmed a HAdV-14 infection.
-
Viral DNA was extracted from Chongqing2024-115 using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Whole-genome sequencing (WGS) was performed by iGeneTech Biotechnology Ltd. using a probe-based hybrid capture approach, followed by next-generation sequencing. This process yielded over 1 Gb of sequencing data, achieving complete genome coverage (100%) at a depth exceeding 8,000×. Sanger sequencing was used to validate genomic regions containing ambiguous bases. The genome was annotated using Geneious Prime software by mapping the sequences to the HAdV-14 prototype strain (de Wit, GenBank accession number AY803294). Complete genomic sequence of the Chongqing2024-115 strain in this study was deposited in the China National Microbiology Data Center with accession number NMDCN0009AD2.
-
All available HAdV-14 WGSs were retrieved from GenBank. After removing incomplete and redundant sequences, a dataset of 67 WGSs from nine countries (1955–2024) was established. This dataset included five Chinese strains (2010–2013) and 62 strains from eight countries and regions (1955–2024), including the United States, Canada, Japan, Honduras, Ireland, Nepal, the Netherlands, and Scotland. With the addition of the newly sequenced strain Chongqing2024-115 from this study, a final dataset of 68 WGSs was compiled for subsequent phylogenetic and genetic variation analyses.
-
Sequence alignment was performed using ClustalW in MEGA v7 (http://www.megasoftware.net), followed by pairwise similarity assessment using BioEdit v7.0.4.1 (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). A maximum likelihood phylogenetic tree was constructed using MEGA v7, with bootstrap support (>80%) indicated at the corresponding nodes. In addition, a phylogenetic network based on WGSs was constructed using SplitsTree v4.19.2 (http://www.splitstree.org) for viral evolutionary analysis. Genetic variations were identified using the Snipit package, focusing on nucleotide mutations at a frequency of 100%. Recombination analysis was conducted using RDP4 (http://web.cbio.uct.ac.za/~darren/rdp.html) and SimPlot v3.5.1 (http://sray.med.som.jhmi.edu/RaySoft/SimPlot) (window size, 2,000 bp; step size, 100 bp).
-
The complete genome of Chongqing2024-115 was 34,766 bp in length, with a GC content of 48.85%, consistent with the previously reported HAdV-14 genome (11). Annotation using the HAdV-14 prototype strain (AY803294) as a reference identified 38 protein-coding regions (
Supplementary Table S1 ). Nucleotide sequence alignment revealed 99.6% identity with the prototype strain. Further analysis of 12 key functional regions, including genes encoding major capsid proteins (penton base, hexon, and fiber), core proteins (pV, pVII, pTP, and pIVa2), minor proteins (pIX, pIIIa, pVI, and pVIII), and the non-structural protein pX, showed that, with the exception of the fiber gene region (nt, 99.1%; aa, 98.7%), the remaining 11 regions exhibited high sequence similarity compared with the prototype strain (nt, 99.6%–100.0%; aa, 99.0%–100.0%) (Table 1).Coding regions Identity % NT AA Major capsid protein penton base 99.8 99.6 hexon 99.8 99.8 fiber 99.1 98.7 Core protein pV 99.9 100.0 PVII 99.8 100.0 pTP 99.6 99.0 pIVa2 99.7 99.2 Minor protein pIX 100.0 100.0 pIIIa 99.8 100.0 pVI 99.8 99.5 pVIII 99.8 100.0 Non-structural protein pX 100.0 100.0 Whole genome sequence 99.6 Abbreviation: NT=Nucleotide; AA=Amino Acid. Table 1. Sequence identity between strain Chongqing2024-115 and the HAdV-14 prototype strain (de Wit, AY803294).
-
To elucidate the genetic relationship between Chongqing2024-115 and previously reported domestic and international HAdV-14 strains, phylogenetic analysis was conducted using the constructed WGS dataset (Figure 1). Consistent with previous findings, all 68 strains clustered into two well-supported clades: the HAdV-14 clade, comprising the prototype strain, and the distinct HAdV-14p1 clade (14). Notably, the HAdV-14p1 clade could be further subdivided into three subclades (I–III; bootstrap values >80%), a finding corroborated by phylogenetic network analysis, which revealed three independent evolutionary trajectories among these strains (
Supplementary Figure S1 ). Nevertheless, all three subclades exhibited high sequence identity (99.5%–99.6%) with the HAdV-14 prototype strain, indicating a highly conserved HAdV-14 genome.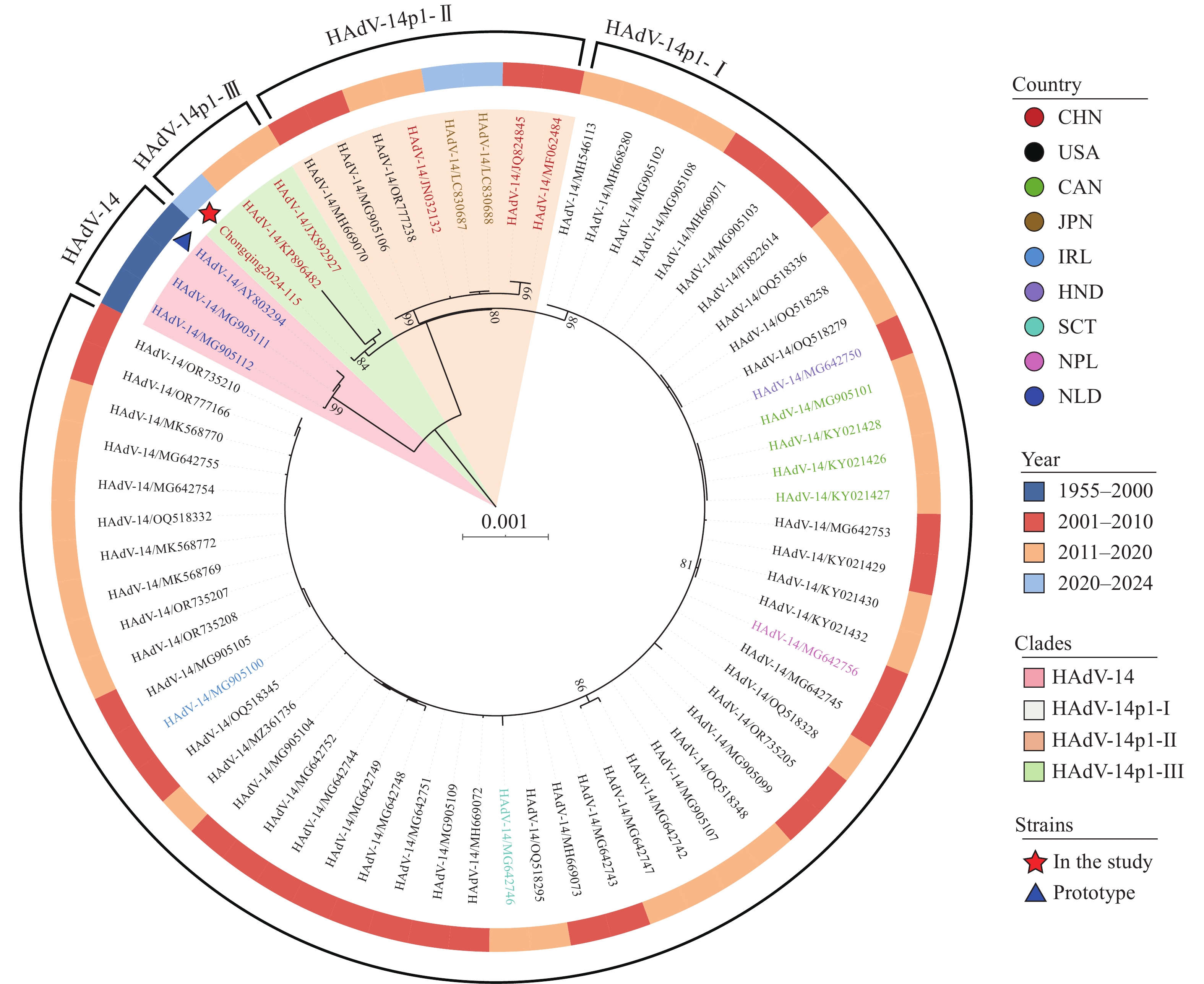 Figure 1.
Figure 1.Maximum-likelihood phylogenetic tree of 68 HAdV-14 whole-genome sequences, including strain Chongqing2024-115 in this study and 67 strains from GenBank database.
Abbreviation: HAdV-14=Human adenovirus type 14.Chongqing2024-115 was phylogenetically assigned to HAdV-14p1 subclade III, which also included two other Chinese strains isolated from Beijing (2012) and Gansu (2013) (intra-subclade genetic distance, 0.00065). In contrast, subclades I and II exhibited broader geographical and temporal distributions. Subclade I comprised 54 strains from six countries, including the United States, Canada, Honduras, Ireland, Nepal, and Scotland (2003–2020; intra-subclade genetic distance, 0.00005). Subclade II consisted of three Chinese strains from Guangzhou (2010) and Beijing (2011), along with five strains from the United States and Japan (2006–2024; intra-subclade genetic distance, 0.00008). High sequence identity was observed among the three subclades (genetic distance, 0.00036–0.00065), indicating a close phylogenetic relationship.
-
WGS-based genetic variation analysis was conducted across HAdV-14p1 subclades I–III using the HAdV-14 prototype strain as the reference genome (
Supplementary Figure S2 ). Comparative genomic analysis revealed that the three subclades shared 99 specific nucleotide variations, comprising 85 single-nucleotide substitutions, six insertions, and eight deletions. These variations were distributed across the entire genome, whereas insertions and deletions were predominantly located in regions 11,000 bp upstream and 31,500 bp downstream. Subclade-specific variations were identified: subclade I harbored four substitutions and one insertion; subclade II contained six substitutions, one insertion, and one deletion; and subclade III possessed three nucleotide substitutions.Detailed analysis of the 12 key functional regions identified 28 shared nucleotide variations across all three HAdV-14p1 subclades. These variations were distributed across 10 protein-coding regions (excluding pIX and pX), resulting in 15 amino acid changes (Figure 2). A shared six-nucleotide deletion (nt751–756) within the fiber gene, located in the knob domain, resulted in the loss of two amino acids (K251 and E252). Two shared nonsynonymous variations in the penton base gene (T983C and G1084A), located within the RGD loop region, caused amino acid substitutions F328S and D362N, respectively. Subclade-specific nucleotide mutations were detected, including a synonymous mutation in the hexon gene (G1341A) specific to subclade I and a synonymous mutation in the pVII gene (G360A) unique to subclade II. Chongqing2024-115 was characterized by two unique nucleotide substitutions: a synonymous mutation (T5601C) in the DNA polymerase gene and a nonsynonymous mutation (G31517A; E250K) within the knob region of the fiber gene (
Supplementary Figure S2 ).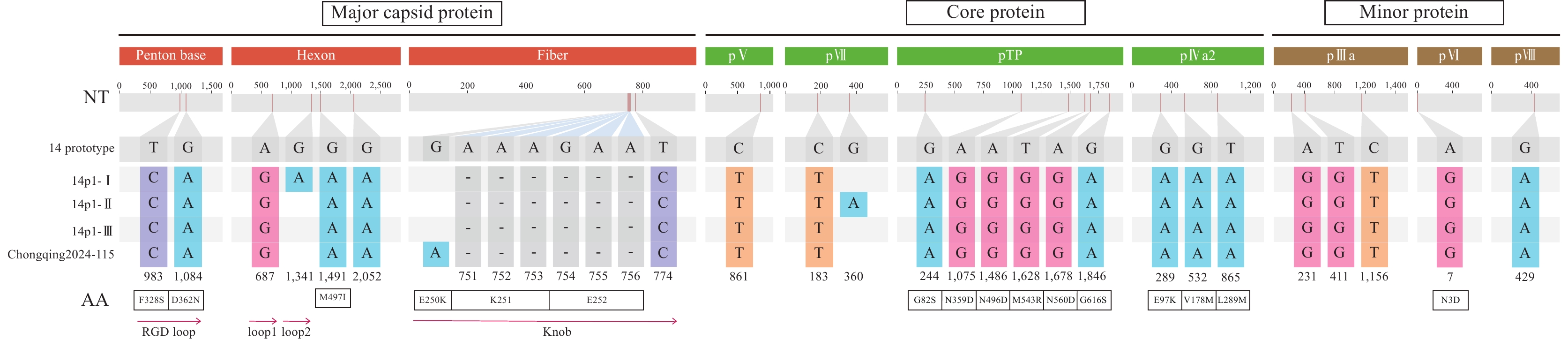 Figure 2.
Figure 2.Nucleotide and amino acid variations specific to HAdV-14p1 clades and strain Chongqing2024-115 in 10 key coding regions compared to the HAdV-14 prototype strain (de Wit, AY803294).
Note: In the schematic representation, major capsid proteins are shown in orange, core proteins in green, and minor proteins in brown. The annotation “NT” denotes nucleotide variation. “-” indicates nucleotide deletion, and the “AA” row displays amino acid variations resulting from mutations. Important functional regions are marked with red arrows. -
A comprehensive recombination analysis was performed on all available HAdV-14p1 strains, including Chongqing2024-115, using WGSs from nine species B prototype strains retrieved from GenBank (recombinant strains were excluded to avoid analytical bias). The results indicated that no intra-species recombination events were detectable within the HAdV-14p1 clade.
-
Since 2019, no cases of HAdV-14p1 infection have been reported in China. As a highly contagious pathogen associated with substantial hospitalization and mortality, understanding the genetic origin of the recently identified HAdV-14p1 is crucial. We performed a comprehensive genomic characterization of the HAdV-14 strain Chongqing2024-115, isolated from a pediatric patient hospitalized with bronchopneumonia. Phylogenetic analysis revealed that this strain clustered closely with earlier HAdV-14p1 subclade III isolates obtained from respiratory infection outbreaks in Beijing (2012) and Gansu (2013) (13). The low intra-subclade genetic distance (0.00065) further supports a common evolutionary origin among these viruses, suggesting sustained transmission of this subclade within China. The absence of reported cases over the past decade may indicate prolonged cryptic transmission of this virus in the population, potentially due to factors such as insufficient surveillance efforts or the failure to systematically capture cases with mild symptoms, leading to a detection gap.
Compared with subclade III, the other two subclades (subclade I, detected across six countries during 2003–2020; subclade II, identified in three countries between 2006 and 2024) demonstrated broader spatiotemporal distributions. These subclades have maintained considerable genomic conservation over nearly two decades, as reflected by their low intra-subclade genetic distance (≤0.00065). All three subclades exhibited high sequence identity (99.5%–99.6%) with the prototype HAdV-14 strain (de Wit, 1955), and the 99 shared variations (85 substitutions, six insertions, and eight deletions) represent a conserved genetic signature of the HAdV-14p1 variant that has circulated globally since its re-emergence after an approximately 50-year absence. Notably, the six-nucleotide deletion in the fiber gene (nt751–756) resulted in a two–amino acid deletion (K251 and E252) in the knob domain. This deletion has previously been identified as a hallmark genetic characteristic distinguishing the HAdV-14p1 strain from the HAdV-14 prototype (14). This two–amino acid deletion has been consistently present in all globally circulating strains identified since 2003, suggesting an important role in the adaptive evolution of HAdV-14p1. Additionally, strain Chongqing2024-115 harbors a unique amino acid substitution (E250K) within the knob domain of the fiber gene. Given the critical role of this domain in mediating viral attachment to and entry into host cells, further investigation is needed to determine whether this substitution affects viral infectivity.
The subdivision of HAdV-14p1 into three distinct subclades underscores the importance of high-resolution genomic data in tracking pathogen evolution. Although the overall genetic distances between subclades were minimal (0.00036–0.00065), consistent clustering patterns suggest that subtle genetic changes may be associated with adaptive evolution or host-specific interactions. The identification of subclade-specific variations and strain-specific mutations, such as those observed in the Chongqing2024-115 isolate, indicates ongoing microevolution within the HAdV-14p1 clade. Despite the detection of subclades II and III in China since 2011, research on HAdV-14p1 remains limited, with only five relevant Chinese genomic sequences (2010–2013) available in public databases. This scarcity of genetic data constrains a comprehensive understanding of the molecular epidemiology and endemic transmission patterns of HAdV-14p1 in China. Therefore, strengthening genomic surveillance is necessary to elucidate the prevalence, evolution, and public health impact of HAdV-14p1 in China.
In conclusion, WGS-based analysis provided a detailed view of the genetic characteristics of the newly identified HAdV-14p1 strain in China in 2024. Our findings confirm that the contemporary HAdV-14p1 strain shares common genetic ancestry with earlier circulating Chinese subclade III strains. Given the association between HAdV-14p1 and severe illness, ongoing surveillance is imperative to gain a deeper understanding of its prevalence and evolution in China and to provide a basis for the formulation of targeted prevention and control measures.
-
The sentinel surveillance hospital in Chongqing for providing the viral strains and associated clinical data. This study used only viral strains and did not involve specimen collection.
HTML
Strain Source and Clinical Information
Genomic Sequencing and Annotation
Dataset
Bioinformatics Analyses
Genomic Characterization
Phylogenetic Analysis
Genetic Variation
Genetic Recombination
| Citation: |

|