

 Download:
Download:




-
Human noroviruses (NoVs) are the leading cause of acute viral gastroenteritis (AGE) worldwide for all age groups (1). NoVs are highly contagious, making them the major cause of outbreaks, especially in semi-enclosed settings such as schools, hospitals, and nursing homes (2). Similar to other RNA viruses, NoVs gain novel genetic properties not only by accumulating mutations that make NoVs evade the host immune response but also by recombination at the junction of the open reading frame (ORF) 1 and 2 that can result in the emergence of a novel strain (3).
Among the more than 30 genotypes of norovirus that cause disease in humans, GII.4 viruses have been reported to cause the majority of norovirus infections worldwide (4). Novel GII.4 variants have emerged every several years, displacing previous dominant strains to cause a new wave of outbreaks (4). The GII.4 Sydney [P31] variant (previously named GII.Pe.-GII.4 Sydney) emerged in late 2012 and became a prevalent strain worldwide (5). GII.4 Sydney [P16] emerged in 2015 and replaced GII.4 Sydney [P31] as the predominant strain in the US and European countries in the 2016–2017 winter season (5).
GII.4 Sydney [P31] emerged in 2012 and caused outbreaks during the 2012–2013 season and has remained the predominant strain in sporadic cases up to the present day in China (6). Although non-GII.4 genotypes GII.17[P17] and GII.2[P16] caused outbreaks and increased in prevalence during the 2014–2015 and 2016–2017 seasons, respectively, GII.4 Sydney [P31] has been continuously detected in outbreak surveillance (7-8). This study described the outbreaks in China caused by GII.4 Sydney [P31] from October 2016 to December 2020, as well as the genome information of GII.4 Sydney [P31], the important amino acid mutation sites of antigen epitopes and receptor binding sites, human histo-blood group antigens (HBGAs), which were assumed to be associated with the epidemic pattern of GII.4 noroviruses.
Since 2016, laboratory-based surveillance of norovirus outbreaks in China (CaliciNet China) has been conducted, which is a network of county-level, city-level, and provincial CDCs coordinated by China CDC. As a representative member of the Caliciviridae family, norovirus has cup-shaped depressions under the electron microscope, which is the origin of the network name. A detailed description of CaliciNet China has been reported previously (8). By 2018, the number of network laboratories has increased to 31, 14 provinces are covered, including 11 provincial-level network laboratories (Beijing, Chongqing, Shanghai, Hebei, Shandong, Jiangsu, Zhejiang, Hubei, Hunan, Fujian, and Guangdong CDC), 11 municipal network laboratories (Shenyang, Qingdao, Jinan, Wuxi, Huai’an, Nanjing, Mianyang, Guilin, Xiamen, Guangzhou, and Shenzhen CDC); and 8 district-level network laboratories (Beijing’s Fengtai, Changping, Mentougou, Shunyi, and Chaoyang Districts, Shanghai’s Changning District, Guangdong’s Huadu District, and Shenzhen’s Nanshan and Luohu District CDC). Norovirus outbreaks were defined as >5 epidemiologically linked AGE cases within 3 days with >2 norovirus positive samples confirmed through real-time reverse transcription-polymerase chain reaction (real-time RT-PCR) (whole fecal, rectal swab, or vomitus) (8). Commercial norovirus real-time RT-PCR kits (BioPerfectus Technology Co., Jiangsu, China; Aodong Technology Co., Shenzhen, China) were used by network labs. Norovirus positive samples were then amplified using the dual polymerase-capsid genotyping protocol (5). The near-complete genome of GII.4 Sydney [P31] strains from 4 different outbreaks were sequenced by Sanger sequencing using primers designed by primer 5.0 (version 5.0, Premier, Palo Alto, Canada) (Supplementary Table S1). Contig assembly was performed by DNAstar (version 7.0, DNASTAR, Madison, Wisconsin, USA) and sequence alignments with Clustal W (version 2.0, Conway Institute UCD Dublin, Dublin, Ireland) (
http://www.clustal.org/clustal2 ). Phylogenetic analysis of these sequences was performed in the MEGA (version 6.0, Mega Limited, Auckland, New Zealand). Phylogenetic trees with bootstrap analysis from 1,000 replicates were generated by using the neighbor-joining method.During October 2016–December 2020, 1,954 norovirus outbreaks were reported to CaliciNet China. The numbers of cases were available in 1,631 outbreaks (83.47%) and ranged from 5 to 391 people per outbreak, with a median of 15 people. Of the 1,887 outbreaks with the reported setting, 852 (45.15%) occurred in childcare centers, followed by 565 (29.94%) in primary schools, 223 (11.81%) in middle schools, and 94 (4.98%) in other schools. Of 1,833 (93.80%) with reported transmission mode, person-to-person (1,748, 95.36%) was predominant, followed by 59 (3.21%) foodborne and 26 (1.42%) waterborne transmission. The season peaks occurred from November–December and in March of the subsequent year (Figure 1).
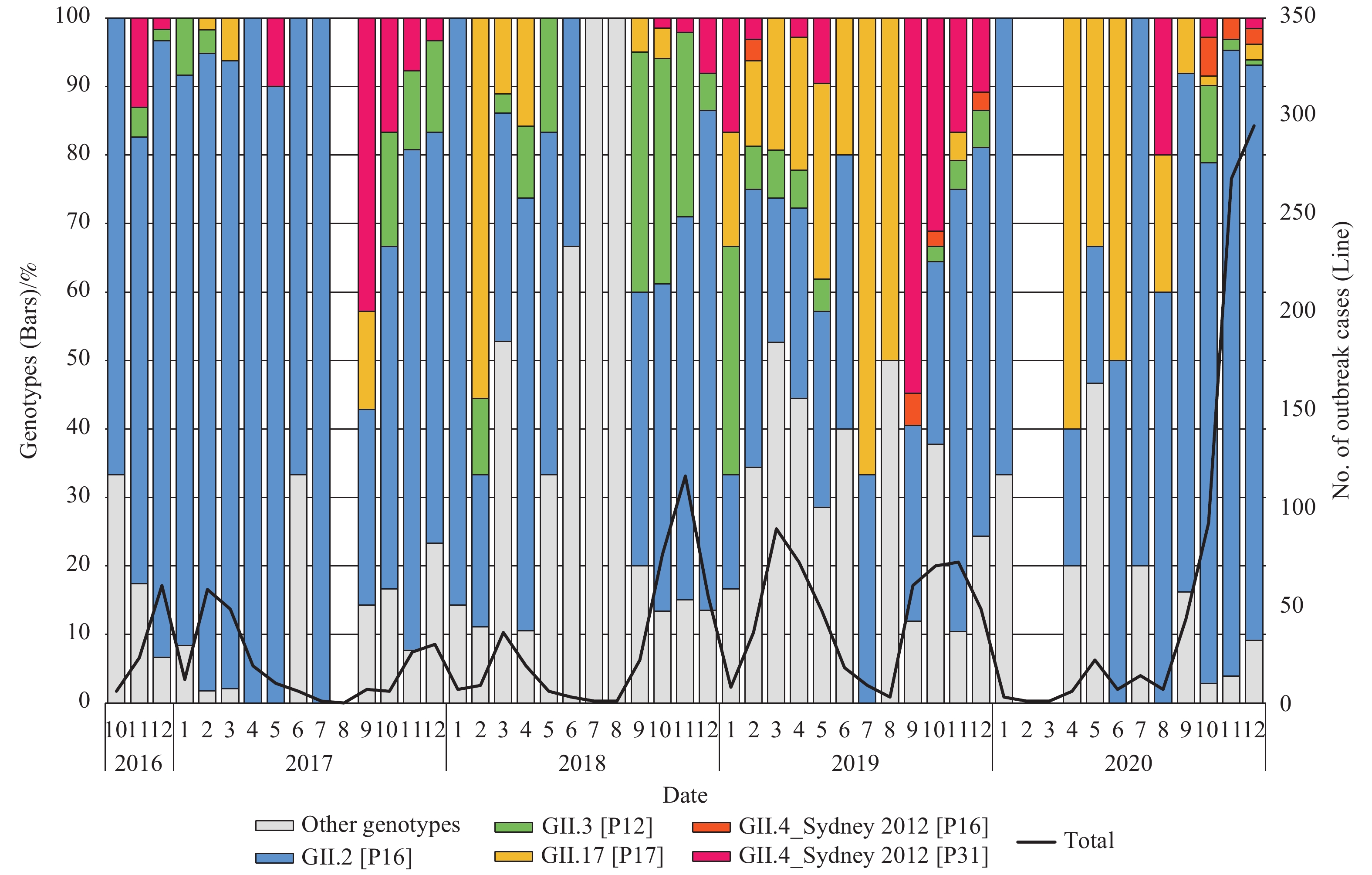 Figure 1.
Figure 1.Monthly trends of norovirus genotypes for outbreak cases of acute gastroenteritis outbreak uploaded to CaliciNet China, October 2016–December 2020.
Among the 1,352 outbreaks (69.19%) that were genotyped, 1,244 cases (92.01%) were GII positive, 63 cases (4.66%) were GI positive, and 45 cases (3.33%) were mixed genotypes. Overall, 10 GI genotypes and 16 GII genotypes were detected. Among the GII outbreaks, 69.0% (859/1,244) were GII.2 [P16], followed by GII.3 [P12] [101 (8.12%)], GII.6 [P7] [82 (6.59%)], GII.4 Sydney [P31] [77 (6.19%)], and GII.17 [P17] [73 (5.87%)]. Other genotypes accounted for less than 5%. The proportion of GII.4 Sydney[P31] in different epidemic seasons of GII outbreak was 2.1% (October 2016–August 2017), 5.5% (September 2017–August 2018), 3.3% (September 2018–August 2019), 26.6% ( September 2019–August 2020), and 1.12% (September 2020–December 2020) (Figure 1).
In the GII.4 Sydney [P31] outbreak, foodborne transmission accounted for 2.60% (2/77), water-borne transmission accounted for 2.60% (2/77), and interpersonal transmission accounted for 87.01% (67/77). GII.4 Sydney [P31] outbreak sites include kindergartens (63.63%, 49/77), primary schools (13.00%, 10/77), hospitals (5.19%, 4/77), families or communities (3.90%, 3/77), nursing homes (2.60%, 2/77), junior high schools (1.30%, 1/77), universities (1.30%, 1/77), other schools (1.30%, 1/77), and companies (1.30%, 1/77) (Figure 2).
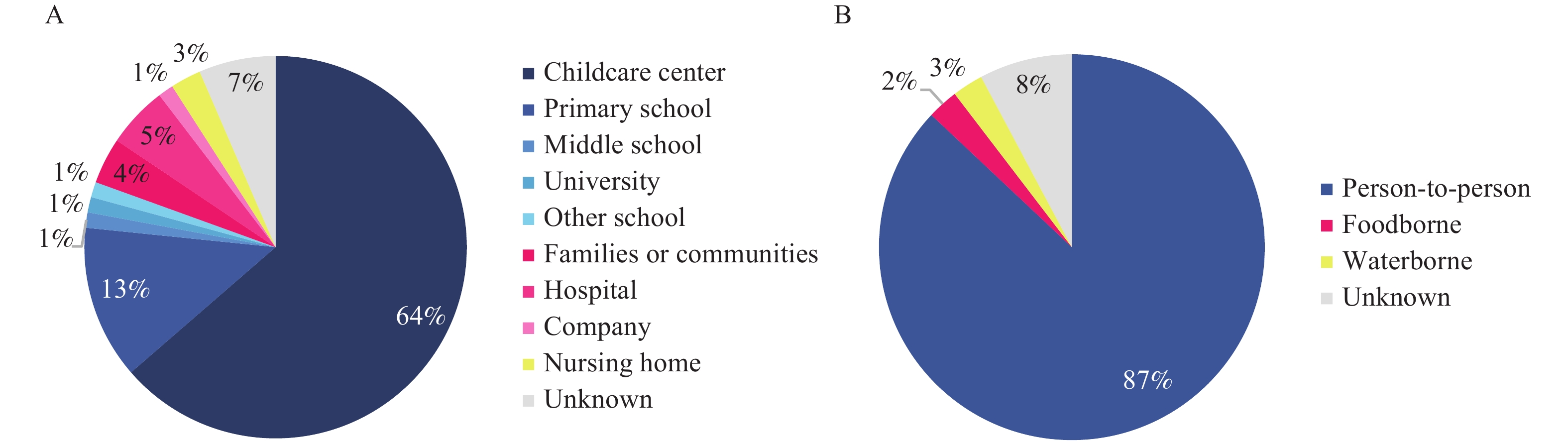 Figure 2.
Figure 2.Settings and transmission routes of GII.4 Sydney[P31] norovirus outbreaks uploaded to CaliciNet China, October 2016–December 2020. (A) the outbreak settings of GII.4 Sydney [P31] norovirus outbreaks. (B) the transmission routes of GII.4 Sydney[P31] norovirus outbreaks.
A total of 9 near-complete genomes of GII.4 Sydney [P31] strain were obtained from 4 outbreaks by Sanger sequencing during 2019–2020 (1 in 2019 and 8 in 2020). Sequence alignment showed that the strain from this study shared a pairwise identity of 95.6%–99.9% at the nucleotide level and 98.3%–100% at the amino acid level. According to the phylogenetic analysis in ORF2, the Sydney 2012 clades can be further divided into 2 subclades, according to the GII.P16 and GII.P31 polymerases. The strains from this study belonged to GII.4 Sydney 2012 [P31] subclades, which showed a general time distribution (2012–2013, 2014–2017, and 2018–2020 clusters). In addition, 2 strains from an outbreak in August 2020 formed a branch independently (Figure 3). The amino acid changes of GII.4 Sydney strain blood group antigen receptor binding site (HBS) tended to be conservative, and only amino acid mutations were found at site II 372 (Figure 4A). Through epitope analysis, the strains after 2014 have changed in epitopes A (297, 372 and 373), B (333), E (414), and H (309 and 310). A total of 4 stains (20HN253–20HN261) from 1 outbreak in December 2020 showed new changes in A epitope (368) and G epitope (355) compared with the previous strains (Figure 4B).
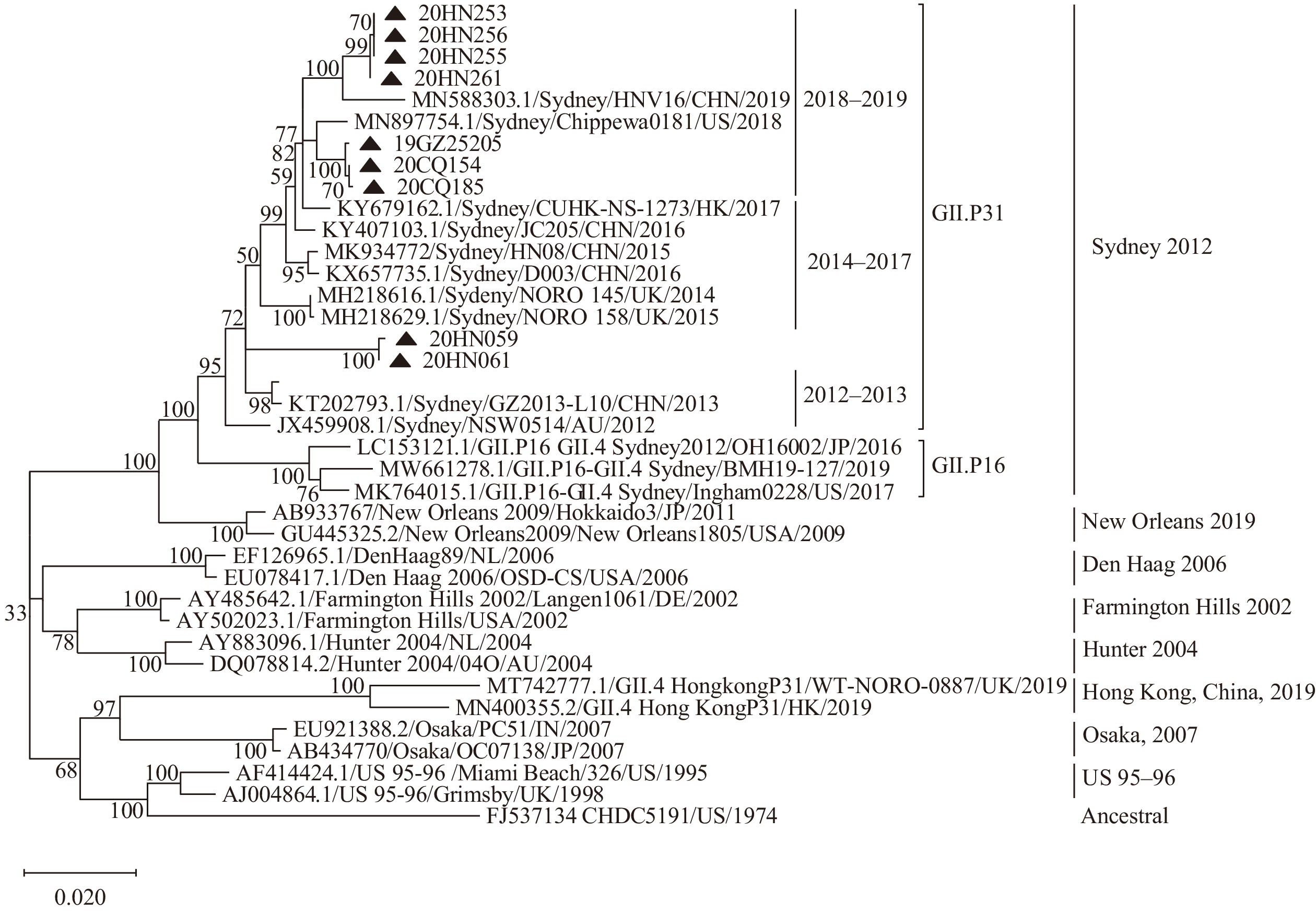 Figure 3.
Figure 3.Neighbor-joining phylogeny of complete sequences of open reading frame 2 of norovirus GII.4 variants.
Note: Strains from this study were marked with a triangle.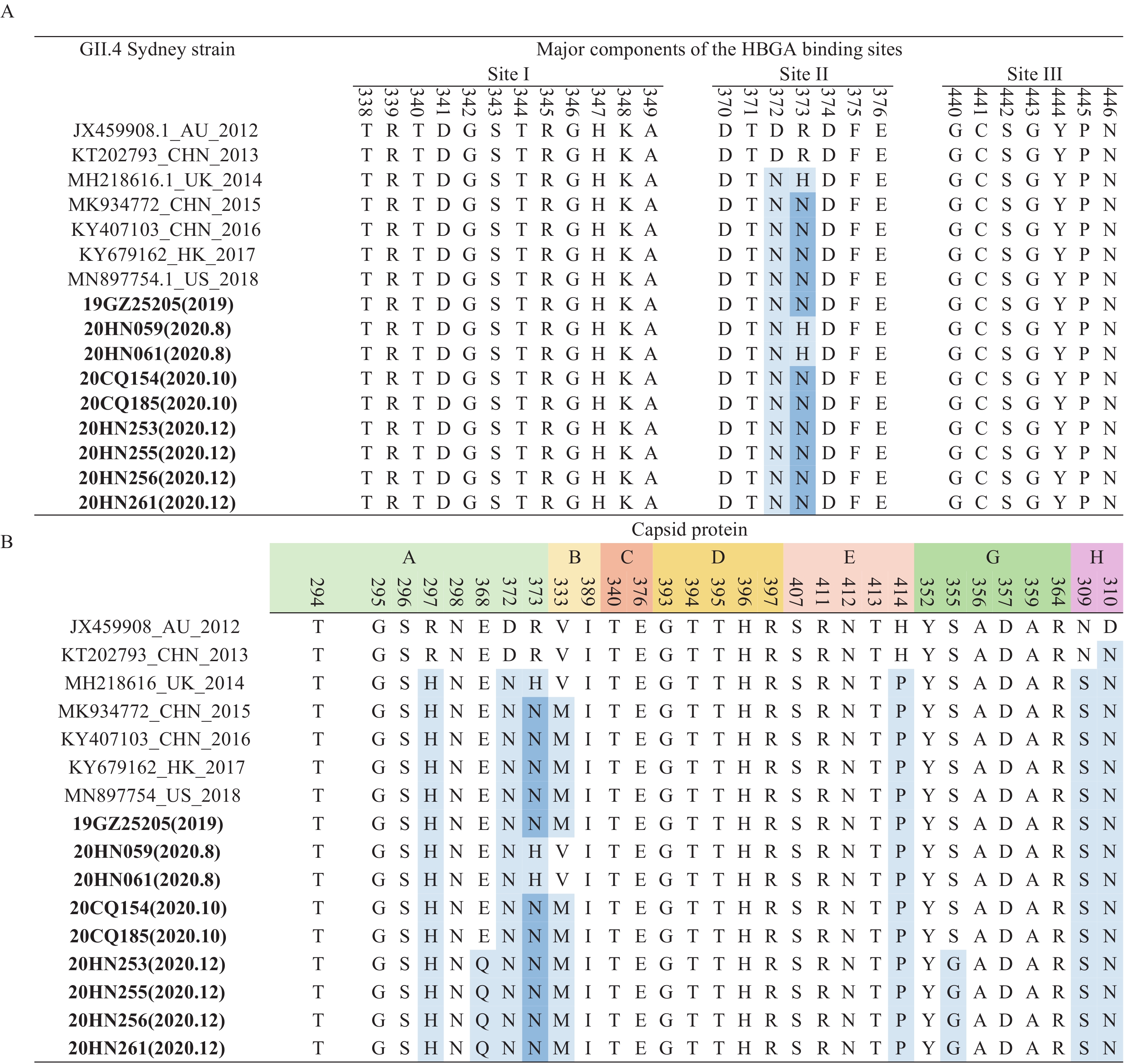 Figure 4.
Figure 4.Sequence analysis of GII.4 Sydney strains. (A) The sequence analysis of histo-blood group antigen (HBGA)–binding interface of the GII.4 major capsid protein VP1 protein. (B) The sequence analysis of antigenic residues of major capsid protein of GII.4 Sydney strains.
Note: Residues mapping on previously characterized A–H epitopes. Amino acid residues that differ from those of the prototype were highlighted in blue. Sequences of this study were in bold. Abbreviations: HBGA=histo-blood group antigen. -
Since CaliciNet China was launched in October 2016 in China, more laboratories were included to report norovirus outbreaks data and sequence information. Therefore, prompt analysis of the genetic characteristics and variation of norovirus strains from China was available. Moreover, the real-time monitoring of the emergence of new norovirus variants has strengthened the capacity on early warning and prediction of norovirus outbreaks in China.
Our study showed the epidemiological characters of outbreaks by noroviruses and identified GII.2 [P16] as the predominant strain in China during October 2016 and December 2020. Similar to studies from other countries in the northern hemisphere, the peak of norovirus outbreaks mainly occurred from October to March of the next year (9). Like other Asian countries such as Japan and the Republic of Korea, schools and kindergartens were also identified as the most common places of norovirus outbreaks in China (10-11). The high proportion of norovirus outbreaks in kindergartens and schools may be related to the high population density in these environments, as well as our enhanced monitoring and reporting of school outbreaks in China (12).
Since mid-1995, the genotype causing the global outbreak of norovirus has only been related to the GII.4 genotype (4). Since 2012, the GII.4 Sydney variant has been predominant all over the world. Therefore, the monitoring of GII.4 Sydney strain is important for potentially emerging GII.4 variants (5). This study described the epidemiological characteristic and the genome variation of GII.4 Sydney [P31]. It was consistent that GII.4 viruses have been associated with person-to-person transmission both from this study and other previous data from CaliciNet USA and NoroNet. The GII.4 Sydney [P31] outbreaks in our study mainly occurred in childcare centers and primary schools. In other parts of the world, GII.4 Sydney viruses have been reported as the dominant genotype among adults and the elderly especially in outbreaks in long-term care facilities (13).
Much evidence from our study suggested that GII.4 Sydney [P31] was undergoing evolution. First, according to the phylogenetic tree, GII.4 Sydney strains with P31 polymerase were clustered in three lineages by epochal evolution (2013–2014, 2014–2017, and 2018–2019) (Figure 3). GII.4 Sydney outbreak strains in this study were mainly clustered in lineage 2018–2019. However, two GII.4 Sydney strains from an outbreak that occurred in 2020 formed an independent branch. Second, compared to reference strains from 2012 to 2013, these GII.4 Sydney strains from 2014–2020 exhibited some changes or switches that occurred in HBGAs binding sites (II) and in epitope sites (A, B, E, G, and H). Notably, GII.4 Sydney outbreak strains in 2020 from our study had unique mutated changes at sites 368 (epitope A) and 355 (epitope G) (Figure 4). Previous studies have confirmed that the amino acid composition of several antigenic epitopes (A, C, D, E, and G) was related to the epidemic pattern of GII.4 variant. A total of 3 epitopes (A, C, and G) contained amino acid residues (352, 357, 368, and 378), which were related to the diversified selection pressure of the new GII.4 variant (14). These residue changes were likely to enable the norovirus to escape the pressure of population immunity and cause a global epidemic of norovirus once again.
In addition to antigenic drift, recombination resulting in polymerase switching was also an important mechanism for the evolution of noroviruses (3). Acquisition of the GII.P16 polymerase and/or associated nonstructural proteins appeared to be the impetus for the predominance of GII.P16-GII.4 Sydney viruses in 2015 to 2016 in the USA (5). However, a limited number of studies have evaluated the evolutionary rate of the ORF1 gene. Each genotype has an evolutionary mechanism. Evolutionary analyses may vary depending on the genotype and the database used (15). Future studies are needed to determine what structural differences contemporary GII.P16 polymerases have gained and what the functional role of these changes is.
There were several limitations in this study. First, local CDCs participated in the network voluntarily, so these data may only represent places covered by Calicinet China and are not generalizable. Still, China CDC is constantly making efforts to include more laboratories and build a more comprehensive surveillance system. Second, the epidemiological information collected in each outbreak was not sufficient, hindering further analysis. Third, the timeliness of the data reporting in CaliciNet China needs to be improved. Due to the increasing burden of laboratory testing caused by COVID-19, not all network members could submit data to China CDC on a monthly basis. In the future, a web-based information submission system will contribute to the near real-time surveillance network.
In conclusion, the molecular epidemiological characteristics of norovirus outbreaks and the genome variation of GII.4 Sydney [P31] from CaliciNet China were analyzed. Data suggested that GII.4 Sydney [P31] had experienced minor but important changes in HBGAs binding sites and antigenic epitope sites associated with the epidemic pattern of GII.4 variant, which should be monitored by continuous surveillance with real-time genotyping and genetic analysis for the emergence of novel GII.4 variant.
-
Jing Wang and all members of CaliciNet China.
HTML
| Citation: |

|