
 Download:
Download:




-
Klebsiella pneumoniae (KP) ranks among the leading causes of healthcare-associated infections worldwide, with high morbidity and mortality driven by carbapenem-resistant and hypervirulent strains The World Health Organization (WHO) listed KP as a critical pathogen on its Bacterial Priority Pathogens List in both 2017 and 2024 (1). Of particular concern are hypervirulent KP (hvKp) and carbapenem-resistant hvKp (CR-hvKp). High-risk clones such as NDM-positive ST147-KL64 (with mortality rates approaching 40%) (2–3) and ST23-KL1 (carrying rmpA/rmpA2, iroB, and/or iucA) are expanding rapidly — at an annual global growth rate of 59% and of 30% in China (4). These trends underscore the urgent need for systematic surveillance of carbapenem-resistant KP (CRKP), hvKp, and CR-hvKp.
Genomic surveillance provides high-resolution genetic insights that enable real-time reconstruction of transmission chains and prospective threat assessment. By elucidating pathogen evolution, dissemination patterns, and clinically relevant phenotypes, it strengthens public health responses (5–6). Clarifying the evolutionary trajectories of antimicrobial resistance (AMR) and virulence determinants further optimizes clinical decision-making and supports targeted infection control (7). This approach represents a paradigm shift from traditional phenotype-based monitoring, enabling outbreak tracing, epidemic forecasting, early warning of high-risk strains, and real-time assessment of emerging AMR and hypervirulence.
Discovering, surveying, and assessing the emergence and spread of hypervirulent KP clones and sequence types requires a comprehensive database of global KP genomes. Although public databases such as the National Center for Biotechnology Information (NCBI) are available, existing resources lack the integration of epidemiological metadata with standardized genomic annotations. To address these critical gaps in global KP surveillance, we established the Klebsiella pneumoniae Genome Database (KPGD), which integrates sequence types, serotypes, antibiotic resistance genes (ARGs), virulence factors (VFs), mobile genetic elements (MGEs), and associated epidemiological metadata — including collection time and geographic origin. KPGD carries significant public health implications by enabling early warning of emerging high-risk clones, supporting outbreak investigations, and assisting public health agencies in monitoring the dissemination of hypervirulent and carbapenem-resistant KP.
HTML
-
KPGD integrates 75,987 KP genomes collected over more than 40 years from 122 countries. Of these, 71,132 publicly available genomes were retrieved from the National Center for Biotechnology Information (NCBI)(8), and 4,855 sequences were collected through national surveillance conducted by the Chinese Pathogen Identification Net (China PIN), which performs hospital-based sampling for febrile respiratory syndrome; KP isolates obtained from lower respiratory tract specimens testing positive by nucleic acid detection were cultured and sequenced. These 4,855 sequences originated from 170 cities across 29 provincial-level administrative divisions (PLADs) in China (
Supplementary Figure S1 ), spanning a period of over 40 years. A major component of the database is the Chinese collection of 17,251 genomes, comprising 12,396 NCBI sequences combined with those from the China PIN. All 75,987 genome assemblies underwent uniform quality assessment using CheckM (Queensland University of Technology, Queensland, Australia, v1.2.2) (9). Genomes with completeness≥95%, contamination≤5%, and consistency≥95% were retained for downstream analysis. -
Genomes in KPGD were curated and analyzed using the Global Catalogue of Pathogens (gcPathogen) frameworks and tools (10–11) (Figure 1 and
Supplementary Material ). Hyper-VFs (iroB, iucA, rmpA, and rmpA2) and resistance genes were manually curated on the basis of a systematic literature review.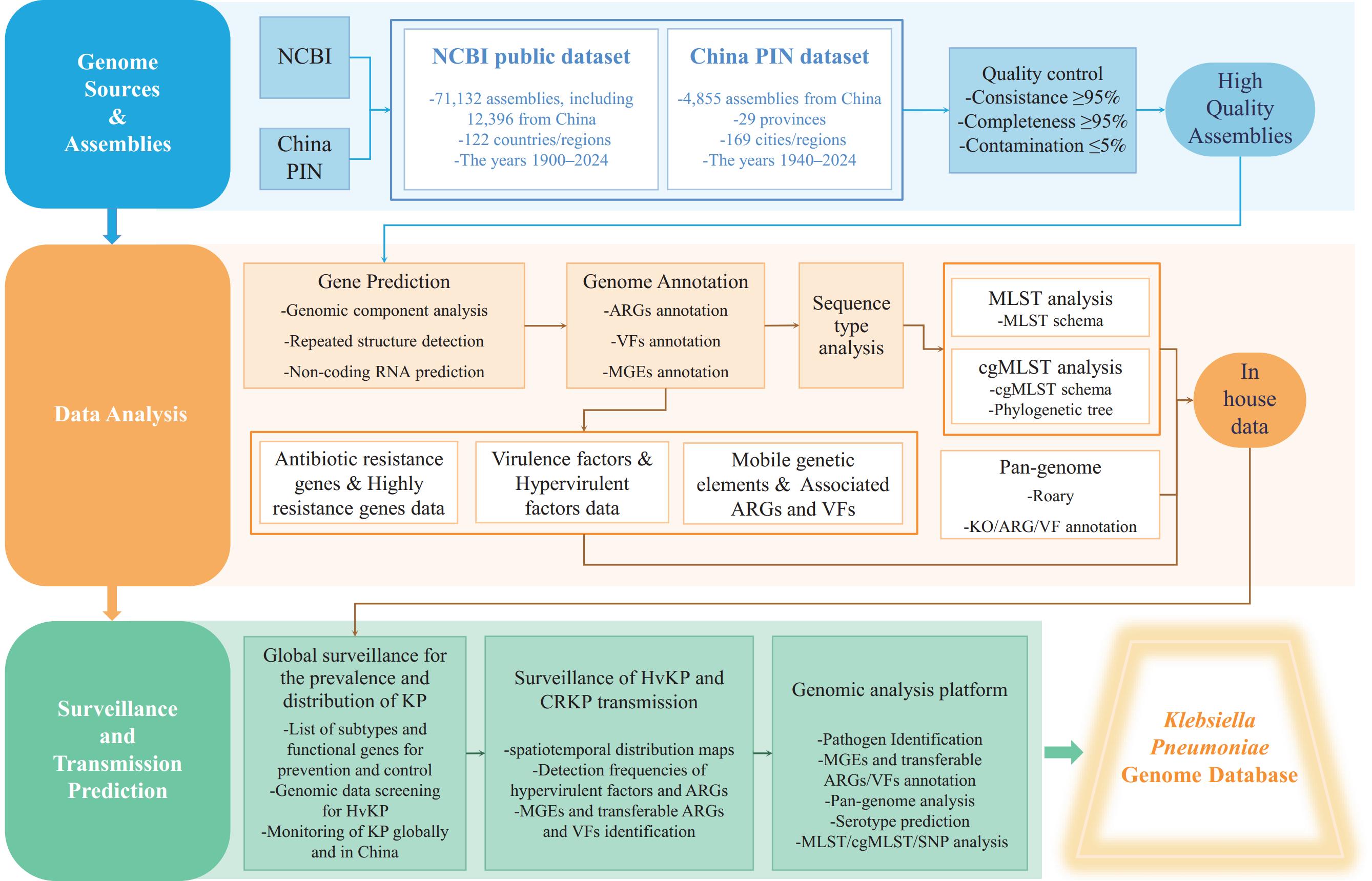 Figure 1.
Figure 1.Data processing pipeline of the KPGD.
Abbreviation: KPGD=Klebsiella pneumoniae Genome Database; China PIN=Chinese Pathogen Identification Net.
Data Source
Analysis Methods
-
The KPGD web platform (Figure 2A) provides comprehensive statistical summaries, including total genome counts and distributions of K/O antigens, hvKp strains, ARGs, sampling countries, and collection years. The "Genome Data" module supports advanced queries by WHO regions, serotypes, sequence types (STs), ARGs, and VFs. Search outputs appear in an interactive tabular format on a secondary page, integrating curated metadata with analytical results. The knowledge graph (Figure 2B) visualizes collaborative networks of research institutions and investigators, along with relevant publications and patents.
 Figure 2.
Figure 2.Features of the KPGD Website. (A) KPGD homepage. (B) The Knowledge Graph page depicting global KP research networks; (C) The hvKp subpage summarizing datasets, spatiotemporal patterns, regional and country-level counts, and ARG/VF frequencies by ST, country, and habitat. (D) Phylogenetic clustering of high-risk KP ST23 clones using the KPGD cgMLST tool.
Note: For (B), it includes authors, institutions, publications, and patents. For (D), the 2019 isolate NMDC60197270 from China PIN clustered within the same phylogenetic branch as isolates from Bangladesh (2016 and 2017), India (2020), and Ghana (2021), suggesting potential transmission from these regions. Furthermore, the 2024 isolate NMDC60192954 clustered predominantly with isolates from Japan (2019–2020), indicating possible transmission from Japan.
Abbreviation: KPGD=Klebsiella pneumoniae Genome Database; China PIN=Chinese Pathogen Identification Net; ST=sequence type.
The AMR module identifies high-frequency, widely distributed ARGs across hosts and ecological niches. The MGE module maps mobile genetic elements harboring critical ARGs and hyper-VFs, enabling assessment of their horizontal transfer potential. The hvKp module captures the dynamics and epidemiological trends of hypervirulent strains (Figure 2C), while the pan-genome analysis module resolves core and accessory gene repertoires, including conserved resistance determinants across lineages.
By integrating these genetic insights with spatiotemporal, host, and clinical metadata, KPGD facilitates early detection of dissemination events, tracks the evolutionary trajectories of resistance and hypervirulence, and monitors the spread of high-risk MGEs — thereby providing a platform for proactive public health intervention. For example, the high-risk clones ST147_KL64_O2a, ST23_KL1_O1ab, and ST45_KL24_O2a were confined to a limited number of countries before 2010 but had spread globally by 2024, demonstrating clear international transmission. Specifically, ST147_KL64_O2a was detected in only 13 countries prior to 2010; its geographic range expanded to 40 countries by 2015 and further to 47 countries by 2024. Similarly, ST23_KL1_O1ab exhibited a pronounced dissemination pattern, with its country-level distribution increasing from 13 countries in 2010 to 32 countries by 2024. Meanwhile, ST45_KL24_O2a was initially confined to 6 countries before 2010, underwent rapid dissemination to reach 28 countries by 2015, and continued cross-border transmission, expanding further to 32 countries by 2024.
Additionally, KPGD offers five integrated one-stop online tools (
Supplementary Material ) for pathogen identification, Single Nucleotide Polymorphism (SNP) analysis, serotype prediction, detection of MGEs and transferable ARGs and VFs, and Core Gene Multilocus Sequence Typing (cgMLST) analysis (Figure 2D). -
Genomic surveillance through KPGD reveals a dynamic and evolving risk landscape characterized by distinct evolutionary trajectories among high-risk KP lineages under different antibiotic selection pressures, as stratified by the World Health Organization (WHO) Access, Watch, Reserve (AWaRe) classification (12) (Figure 3A). These findings underscore the need for lineage-specific surveillance and intervention strategies.
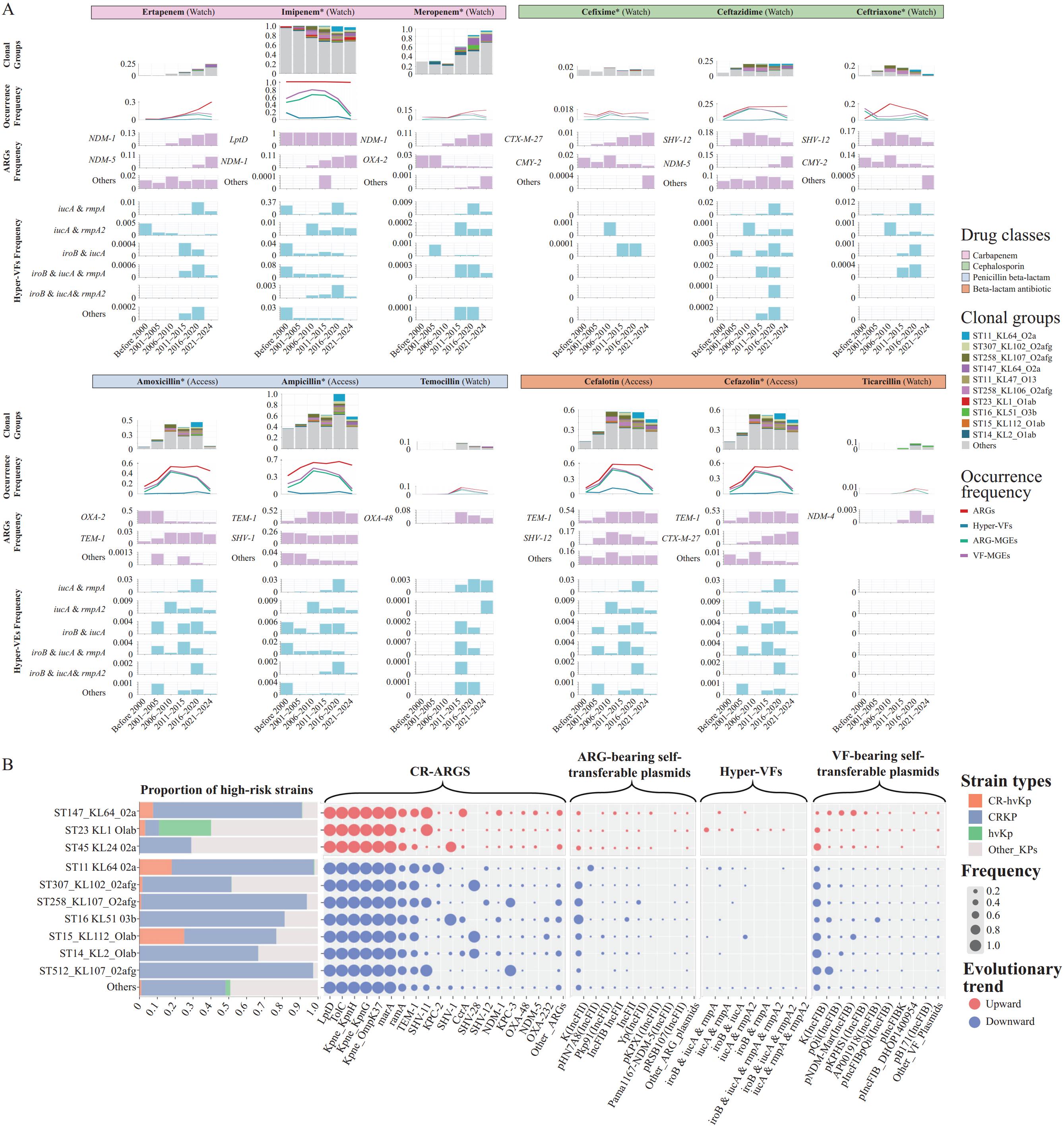 Figure 3.
Figure 3.Risk Profiles and Evolutionary Trajectories of KP. (A) Longitudinal trends in ARGs, hyper-VFs, and major clonal lineages among KP isolates (before 2000–2024). (B) Risk assessment of the top 10 clonal lineages based on resistance and virulence determinants.
Note: For (A), Antibiotics are classified according to the WHO AWaRe framework ("Access" and "Watch"). Stacked bars represent the frequencies of the top 10 clonal lineages (with "Others" denoting remaining types). Line plots depict temporal changes in overall resistance and virulence frequencies under antibiotic pressure, while area charts summarize the occurrence of key ARGs and hyper-VFs. For (B), Bars represent different clonal groups, with colors indicating high-risk clones, including CR-hvKP, CRKP, and hvKP. Bubble plots illustrate the frequencies of carbapenem-resistance and hypervirulence genes, as well as the frequencies of self-transferable plasmids associated with these determinants. Red bubbles indicate an increasing trend for the corresponding clonal group, whereas blue bubbles indicate a decreasing trend.
Abbreviation: WHO=World Health Organization.
Clonal group analyses identified several high-risk lineages (Figure 3A), notably ST11_KL64_O2a and ST258_KL107_O2afg, detected at elevated frequencies (peaking at around 0.2, particularly after 2016) under "Watch" antibiotic selection (carbapenems and other β-lactams). ST11_KL64_O2a persisted from 2010 to 2024 with pronounced enrichment among carbapenem-resistant isolates, indicating adaptation to last-resort therapies. ST307_KL102_O2afg peaked among carbapenem-resistant strains before 2015 (>0.07), potentially reflecting increased use of imipenem and meropenem during that period (13), before declining rapidly to below 0.1. ST147_KL64_O2a exhibited marked post-2020 expansion (around 0.04), especially among carbapenem- and ampicillin-resistant isolates. The hypervirulent lineage ST23_KL1_O1ab remained at low frequencies (0–0.05), although its recent increase among imipenem-resistant isolates suggests progressive acquisition of resistance to broad-spectrum β-lactams.
At the population level, resistance frequencies displayed distinct temporal patterns. Carbapenem-resistance ARGs increased steadily, with β-lactam ARGs among ertapenem- and meropenem-resistant isolates rising from near zero in 2010 to 0.15–0.3 by 2024. In contrast, cephalosporin resistance declined overall (below 0.25), while penicillin β-lactam resistance fluctuated between 0.1 and 0.7. These trends underscore a shifting AMR landscape and highlight lineage-specific evolutionary strategies under defined antibiotic pressures, illustrating the adaptive diversification of high-risk KP clones in response to therapeutic interventions.
Temporal dynamics of key carbapenem-resistance ARGs ("Watch" antibiotics) further emphasize this expansion (Figure 3A). NDM-1 emerged at 0.038 (2010–2015) and peaked at 0.13 (2022–2024). NDM-5 rose from undetectable levels to 0.11 by 2022–2024. In contrast, ARGs associated with "Access" antibiotics (e.g., TEM-1 in amoxicillin/ampicillin-resistant isolates) remained stable (around 0.1–0.5), consistent with their role in first-line therapy. Other ARGs (OXA-2, CMY-2, and CTX-M-27) persisted at low prevalence. Notably, the near-universal presence of LptD (frequency≈1.0) among imipenem-resistant isolates suggests a potential role in KP survival under carbapenem pressure. These ARGs were predominantly disseminated by conjugative IncFII-type plasmids, including K(IncFII), pHN7A8, and pKP91, whose frequencies fluctuated widely (0 to 0.6), underscoring their central role in the rapid horizontal transfer of carbapenem resistance.
Hyper-VF diversity was closely associated with resistance profiles across KP populations (Figure 3A). Among imipenem-resistant isolates ("Watch" antibiotic), hyper-VF richness was pronounced. Before 2000, the dominant combination (iroB, iucA, and rmpA) peaked at 0.08 and then declined steadily. Between 2011 and 2020, a two-factor combination (iucA and rmpA) predominated (around 0.04), surpassing all others. By 2024, all hyper-VF combinations had decreased to around 0.005. These hyper-VFs were largely disseminated by self-transmissible IncFIB-type virulence plasmids, including K(IncFIB) and pQil, peaking at around 0.5.
As shown in Figure 3B, ST23_KL1_O1ab harbored a substantially higher proportion of hvKP strains than other lineages. CR-hvKP strains were most frequently detected in ST15_KL2_O1ab, ST11_KL64_O2a, and ST147_KL64_O2a. Notably, the three clonal lineages displaying an increasing trend carried relatively low frequencies of ARG-bearing self-transferable plasmids. In contrast, all other clonal lineages (with decreasing trends) carried the ARG-bearing self-transferable plasmid K(IncFII), with the highest frequency exceeding 0.8. However, all lineages carried the VF-bearing self-transferable plasmid K(IncFIB), with a maximum frequency of approximately 0.6.
Time-series analyses revealed that fluctuations in virulence plasmid prevalence closely aligned with antibiotic selection regimes. Higher plasmid frequencies were associated with "Access" antibiotics, particularly first-generation cephalosporins (e.g., cefalotin and cefazolin). This suggests that virulence plasmid dissemination may be favored under first-line antibiotic pressure, potentially compensating for resistance-associated fitness costs and enhancing KP persistence.
Overview of the KPGD Web Interface and Analytical Modules
Temporal Dynamics and Evolutionary Trends of High-Risk KP Lineages
-
This study establishes the Klebsiella pneumoniae Genome Database (KPGD) as a global resource for genomic surveillance, addressing the public health threats posed by the dissemination, pathogenicity, and antimicrobial resistance of hypervirulent and antibiotic-resistant lineages. By integrating 75,987 genomes from 122 countries with epidemiological metadata and standardized bioinformatic pipelines, KPGD enables high-resolution tracking of KP transmission, evolution, and risk patterns across decades and continents. These datasets can also be integrated into national monitoring systems, such as the China PIN, to support epidemic analysis, risk assessment, and early warning of high-risk KP clones.
Genomic surveillance revealed distinct evolutionary trajectories among high-risk lineages under differential antibiotic selection pressures. Clones including ST11_KL64_O2a and ST258_KL107_O2afg persisted under carbapenem-dominated "Watch" antibiotics, driven by horizontal acquisition of carbapenemase genes (e.g., NDM-1 and NDM-5) via conjugative IncFII-type plasmids. Temporal fluctuations in plasmid detection correlated with changes in antibiotic usage, underscoring their role as vectors for resistance dissemination.
The expansion of NDM-positive ST147_KL64_O2a represents a specific example of a high-risk clone in which carbapenem resistance and hypervirulence markers co-occur. This does not imply that NDM universally drives hypervirulence. Future studies should investigate region-specific associations between carbapenemase genes and hypervirulence markers.
Concurrently, a dynamic interplay between resistance and pathogenicity was observed. Under first-line "Access" antibiotic pressure, KP strains gained a selective advantage through acquisition of IncFIB-type virulence plasmids, facilitating the emergence of clones exhibiting both high pathogenicity and antimicrobial resistance (14).
Collectively, these findings provide a genomic framework for guiding public health strategies. KPGD serves as a foundational global resource for identifying emerging high-risk clones, elucidating the molecular drivers of their success, and tracking the horizontal transfer of ARGs and VFs worldwide. Ensuring equitable access to such genomic platforms will be essential for coordinated international responses to the growing threat of hypervirulent and antimicrobial-resistant KP.
To this end, KPGD is an open and freely accessible web platform available at http://nmdc.cn/gcpathogen/kp. Unlike static genomic collections or one-time analytical studies, KPGD provides interactive real-time querying and integrated analytical modules for the global research community without registration or payment. Users worldwide can filter genomes by WHO region, serotype, ST, ARG, VF, collection time, and geographic origin.
| Citation: |

|