
 Download:
Download:




-
Hand, foot, and mouth disease (HFMD) and herpangina (HA) are acute, highly contagious infectious diseases that predominantly affect children under five years of age. Coxsackievirus A4 (CVA4) is an important etiological agent of both conditions (1). As a member of the Enterovirus genus within the Picornaviridae family, CVA4 possesses a single-stranded positive-sense RNA genome of approximately 7.4 kb. The VP1 protein contains critical antigenic determinants involved in viral adsorption and host cell infection, and sequence variability in VP1 forms the basis for enterovirus serotype and genotype classification (2).
The prototype CVA4 strain (AY421762/High Point/1948) was first isolated in 1948 from urban sewage collected during a poliomyelitis outbreak in North Carolina, USA (3). Recent surveillance data indicate an increasing detection rate of CVA4 among patients with HFMD and HA, which correlates with its involvement in large-scale outbreaks across several Chinese provinces, including Jiangsu (4) and Beijing (5), as well as in other countries, such as Thailand (6) and Vietnam (7). CVA4 infection has been associated with severe neurological complications, suggesting that its pathogenic potential and epidemic risk may have been underestimated (8).
Previous studies have indicated that the prevalence of enteroviruses may be associated with climatic factors, such as temperature and humidity (9). The Xizang Autonomous Region has a unique climatic environment, and surveillance for CVA4 remains limited. Thus, the prevalence of CVA4 may have been underestimated. Clinical cases typically represent only the tip of the iceberg of viral transmission. Most transmission is driven by asymptomatic or paucisymptomatic infections among healthy children, particularly infants and young children, who serve as primary reservoirs and vectors for persistent viral circulation within communities (10–11). This study analyzed CVA4 isolates collected from healthy children in the Xizang Autonomous Region between 1996 and 2024 to elucidate the phylogenetic characteristics and epidemiological patterns of this virus in high-altitude regions.
-
A total of 2,091 fecal specimens were collected from healthy children across seven prefecture-level divisions of the Xizang Autonomous Region: Ngari Prefecture, Qamdo, Lhasa, Nyingchi, Nagqu, Xigaze, and Shannan. All specimens were stored at 4 °C and transported to the National Laboratory for Poliomyelitis for further processing. Upon arrival, the samples were processed according to the Poliomyelitis Laboratory Manual (fourth edition), followed by viral isolation in human rhabdomyosarcoma and laryngeal epidermoid carcinoma cell lines.
Viral nucleic acids were extracted from the cell cultures using a Tianlong Ex-DNA/RNA Virus kit (CDC/T327, Tianlong, Xi’an) and the GeneRotex96 nucleic acid extraction instrument (Tianlong, Xi’an). The extracted nucleic acids were subjected to enterovirus-specific real-time reverse transcription polymerase chain reaction (real-time RT-PCR) using the One Step PrimeScript™ RT-PCR Kit (Perfect Real Time; TaKaRa, Dalian, China).
-
Real-time RT-PCR-positive samples were used to amplify the enterovirus VP1 coding region using the PrimeScript™ One Step RT-PCR Kit Ver.2 (TaKaRa, Dalian, China). The 25 µL reaction mixture consisted of 5 µL RNA template and 20 µL master mix, with primers E486/E488 (Enterovirus alpha-coxsackie universal primers), E490/E492 (Enterovirus beta-coxsackie universal primers), and E494/E496 (Enterovirus coxsackie-polio universal primers) (12). PCR products were sequenced by Sangon Biotech Co., Ltd. (Shanghai, China). The resulting AB1 files were assembled and edited using Sequencher software (version 5.4.5; GeneCode, Ann Arbor, MI, USA), and consensus sequences were subjected to a Basic Local Alignment Search Tool on the National Center for Biotechnology Information platform for enterovirus serotype determination.
Viral isolates confirmed as CVA4 were subjected to amplification of the full-length VP1 region (915 bp) using the primers forward CVA4-VP1-F: 5’-ACACGCCGAACGAAGCTAAT-3’ and reverse CVA4-VP1-R: 5’-TTATGTGTGGCTAGATGGCG-3’. Amplification was performed as previously described (13), and the full-length VP1 sequences of all CVA4 strains were assembled using Sequencher software.
-
The VP1 sequences obtained and 27 reference strains representing various CVA4 genotypes (5) were aligned using MEGA (version 12.1, freely available) (14). A maximum likelihood (ML) phylogenetic tree was constructed in MEGA using the optimal model identified by jModelTest (version 2.1.10, freely available), with branch support assessed via 1,000 bootstrap replicates. Genetic distances between sequences were calculated using MEGA and visualized as a heatmap using the corrplot package in R software (version 4.0.5; R Foundation for Statistical Computing, Vienna, Austria).
The phylogenetic tree was analyzed using TempEst software (version 1.5.3, freely available) via root-to-tip regression to assess temporal signals. Aligned sequences were imported into BEAUti, specifying a strict molecular clock and Bayesian SkyGrid coalescence model. The MCMC chain length was set to 1,000,000,000 generations, and a BEAST control file was generated. Phylodynamic inferences were performed using BEAST X software (version 10.5.0, freely available) (15), with convergence confirmed using Tracer software (v1.7.1; effective sample size >200). A maximum clade credibility (MCC) tree was generated using TreeAnnotator (10% burn-in) and visualized using FigTree. SpreaD3 (version 0.9.7, freely available) was used to calculate Bayes factors (BFs) (16), and CVA4 migration routes within the Xizang Autonomous Region were reconstructed based on BF values (BF≥3, posterior probability >0.5).
-
This study analyzed 66 asymptomatic CVA4 carriers identified through surveillance by the Xizang Autonomous Region Center for Disease Control and Prevention between 1996 and 2024 (Figure 1A). The number of carriers exceeded five in 6 years (2000, 2011, 2018, 2020, 2023, and 2024), with 41 cases (62.12%) reported since 2018. Among them, 25 were male and 38 were female; sex information was missing for three cases (Figure 1B). Most carriers were children aged 0–5 years (Figure 1C), predominantly from Lhasa and Xigaze, with more than 20 cases identified in each region (Figure 1D).
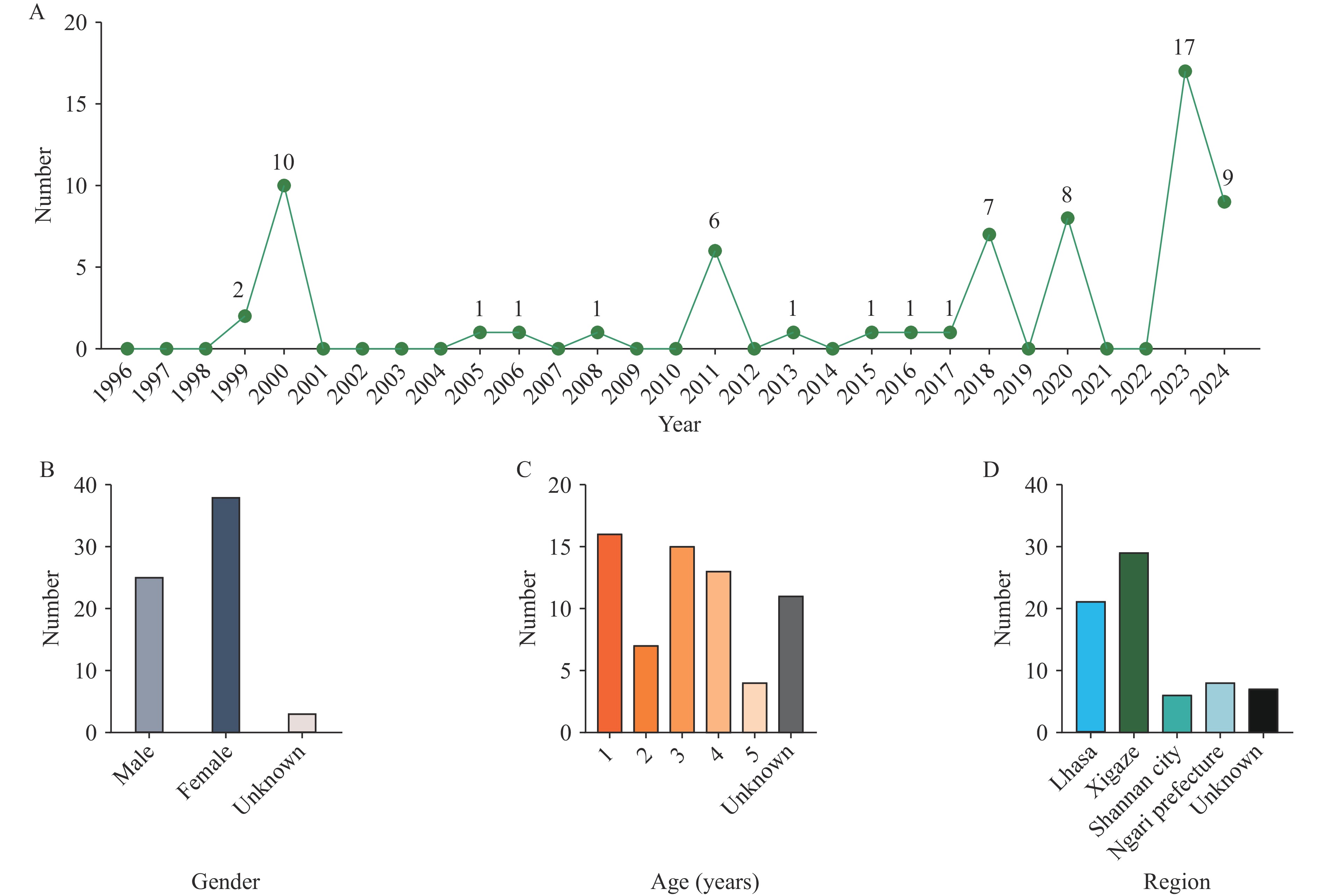 Figure 1.
Figure 1.Demographic characteristics of CVA4-positive healthy carriers. (A) Annual detection count of CVA4 healthy carriers. (B) Sex distribution of carriers. (C) Age distribution of carriers. (D) Geographic distribution of carriers.
Abbreviation: CVA4=Coxsackievirus A4. -
A phylogenetic tree was constructed based on the full-length VP1 sequences from the 66 CVA4 strains obtained in this study and 27 reference strains (Figure 2A). CVA4 segregates into five genotypes (A–E), with genotype D split into sub-genotypes D1 and D2. Genotype A represents the prototype strain (AY421762/High Point/1948); B corresponds to the 1999 Kenyan strain; C consists entirely of Chinese isolates; D1 includes Chinese and Russian strains; D2 comprises Indian, U.S., and Chinese strains; and E contains Chinese and Russian strains. In the Xizang Autonomous Region, genotype C was predominant (56/66, 84.85%) followed by genotype D2 (10/66, 15.15%). Genotype C has been circulating continuously since it was first detected in 1999. The 56 genotype C strains formed two distinct phylogenetic branches: branch 1 contained mainly the 1999–2000 strains, whereas branch 2 consisted almost entirely of strains isolated after 2011. A heatmap of genetic distances confirmed a marked increase between the 2011–2024 sequences and earlier sequences (Figure 2B). Genotype D2 was first identified in Xizang in 2011, with sporadic cases reported in 2015 and 2017, culminating in a clustered outbreak in 2018.
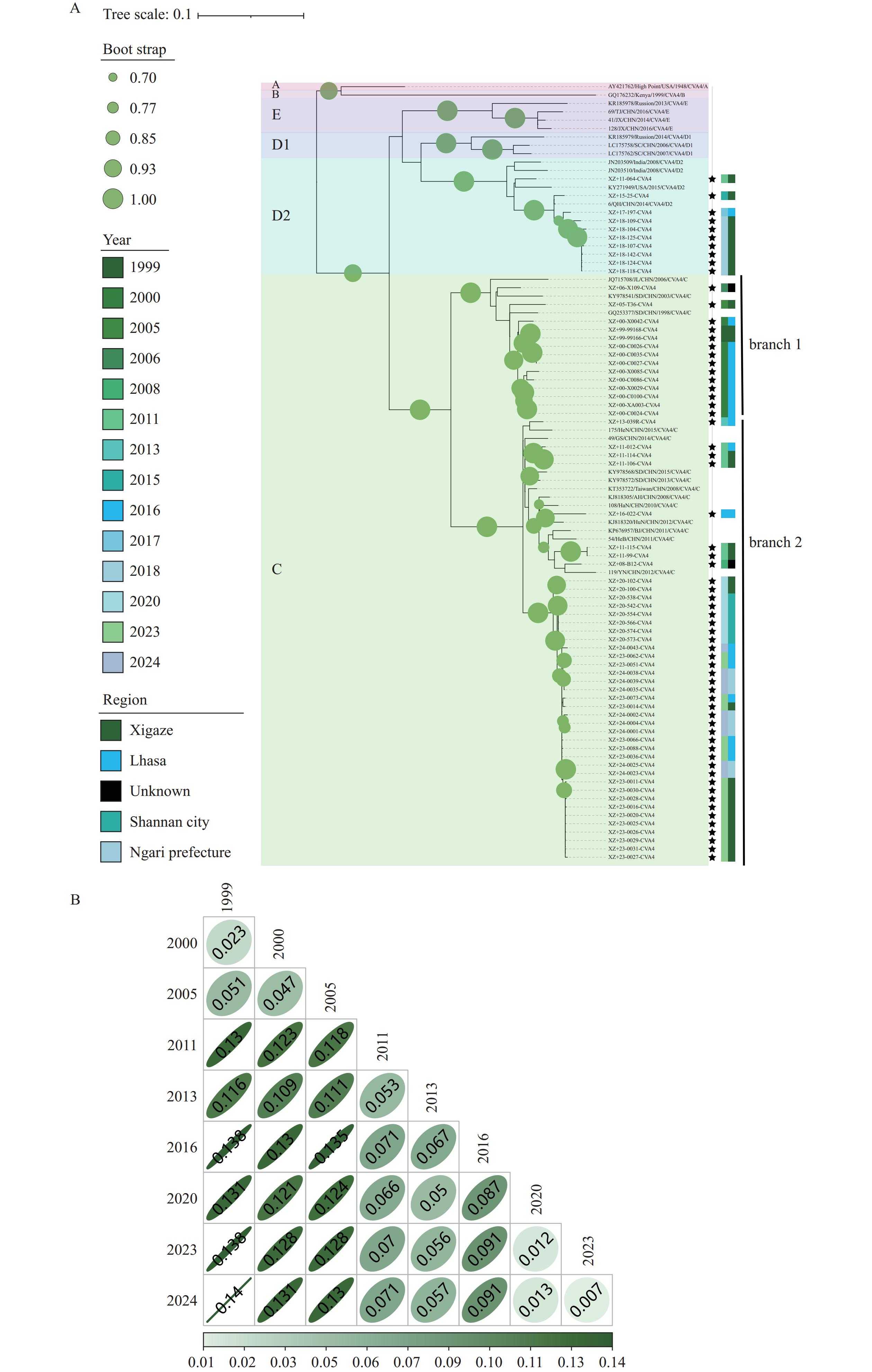 Figure 2.
Figure 2.Phylogenetic analysis of Coxsackievirus A4. (A) Maximum likelihood phylogenetic tree of 66 Xizang CVA4 strains and 27 reference strains. (B) Heatmap of genetic distances among genotype C sequences grouped by collection year.
Note: For panel A, Genotypes A–E are indicated by colored backgrounds; stars denote Xizang strains. The first color-coded block shows collection year; the second block shows sampling region.
Abbreviation: CVA4=Coxsackievirus A4.
-
Analysis of the MCC tree showed strong geographical clustering of CVA4 sequences. In the Xizang Autonomous Region, the virus diverged into genotypes C and D around 1977, consistent with the ML tree (Figure 3A). Genotype C splits into two branches. Branch 1 contained early strains that circulated mainly in Lhasa and Xigaze, with inferred transmission from Lhasa to Xigaze from 1997 to 1999. Branch 2 consisted of strains isolated after 2011, showing repeated cycles of transmission between Lhasa and Xigaze. Subsequent transmissions included Xigaze to Shannan (2018–2020), which led to a local cluster in 2020; Xigaze to Lhasa (2021–2023); and Lhasa to Ngari Prefecture (2022–2024). The D2 sub-genotype circulated only between Lhasa and Xigaze until its disappearance in 2018.
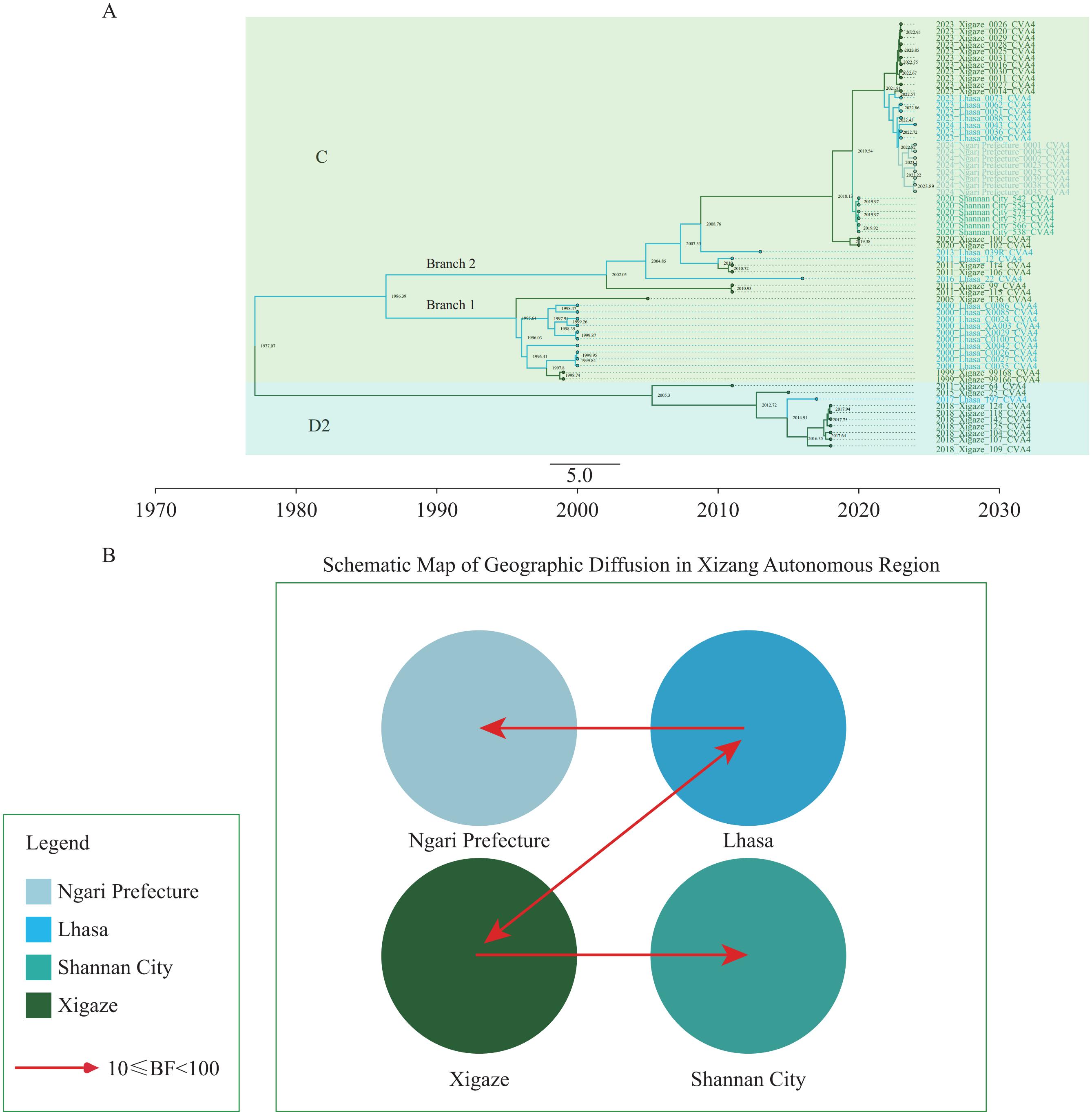 Figure 3.
Figure 3.Geographical transmission pattern of CVA4. (A) Maximum clade credibility tree of 64 Xizang CVA4 sequences. (B) Transmission pathways inferred from SpreaD3 analysis; paths shown have pp>0.5 and BF≥3.
Abbreviation: CVA4=Coxsackievirus A4; pp=posterior probability; MCC=maximum clade credibility; BF=Bayes factor.In the SpreaD3 analysis, transmission routes with a posterior probability (pp) >0.5 and a BF≥3 were defined as well-supported. Based on these criteria, four credible transmission pathways were identified (Figure 3B).
-
As an important member of the enterovirus alphacoxsackie family, CVA4 is one of the most frequently detected coxsackievirus types in non-polio enterovirus surveillance (17). The synthesis of long-term CVA4 surveillance data from the Xizang Autonomous Region is crucial for clarifying local epidemiological patterns. Based on VP1 sequence analysis, this study confirmed that genotypes C and D were the dominant genotypes circulating in the Xizang Autonomous Region from 1996 to 2024, consistent with major CVA4 genotypes prevalent in other regions of China (13,18). The disappearance of the D2 sub-genotype during transmission suggests that genotype C may be advantageous in terms of viral fitness, transmissibility, or immune evasion. These advantages have enabled it to outcompete other genotypes and establish a stable lineage. Consequently, continuous monitoring is essential for detecting future shifts in dominant genotypes.
This analysis revealed a significant increase in the genetic distance between genotype C sequences before and after 2011. Following the 2000 outbreak, detection rates remained very low for approximately 10 years, indicating a possible surveillance gap. The 2011 epidemic strain may not be a direct descendant of earlier local strains but rather a newly introduced external variant, which could explain the observed phylogenetic divergence. Notably, the CVA4 C genotype circulating since 2011 has formed three distinct temporal clusters (2020, 2023, and 2024) on the MCC tree with short intra-cluster branches, indicating multiple localized outbreaks across the Xizang Autonomous Region. All inferred transmission routes are strongly supported by BF values (19).
As both a recipient of the virus from Xigaze and a source of its spread to Ngari, Lhasa has functioned as a key transmission hub, reflecting its central political and transportation roles. Repeated outbreaks underscore the considerable transmissibility of the post-2011 CVA4 C genotype among healthy children and highlight the need for continued surveillance. Despite the cold, high-altitude setting and sparse population, the CVA4 C genotype achieved sustained transmission, producing several discrete temporal clusters. This suggests that environmental factors alone do not halt viral spread, and that population mobility and child congregation settings are the principal drivers of outbreaks. Although this study did not analyze the full genomic characteristics of CVA4, it emphasizes the need to strengthen routine preventive measures and establish a molecular surveillance-based early warning system to reduce the risk of future large-scale CVA4 epidemics driven by asymptomatic transmission.
This study conducted surveillance among healthy children and found an increasing trend in CVA4 detection rates, indicating that the virus poses an epidemic risk in the Xizang Autonomous Region. Major transportation hubs are at particular risk of sustained transmission. These findings highlight the need to strengthen local surveillance systems and implement proactive intervention measures to prevent and control CVA4 spread. CVA4 surveillance should be incorporated into the national notifiable disease surveillance system for patients with HFMD to enable timely detection and response.
-
Approved by the Ethical Committee for Life Science and Medical Research Involving Human Subjects, National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention (Approval No: IVDC2024-012).
HTML
Sample Collection and Virus Identification
Molecular Typing of Enteroviruses and Sequencing of the CVA4 VP1 Coding Region
Bioinformatic Analysis
Demographic Characteristics of CVA4-positive Healthy Carriers
Phylogenetic Analysis Based on CVA4 Full-length VP1 Sequences
Geographical Transmission Pattern of CVA4
| Citation: |

|