
 Download:
Download:




-
Introduction: To investigate transcriptional differences and their implications for evolutionary relationships among Bartonella species from diverse host origin.
Methods: Illumina high-throughput sequencing technology was used to sequence the transcriptomes of eighty-seven Bartonella strains. The differences in gene expression among strains from different species and hosts were analyzed, and the results of the genome phylogenetic analysis were compared to explore the role and influencing factors of transcription levels in Bartonella species differentiation.
Results: The transcriptomes of Bartonella strains varied systematically by species and host origin, with numerous differentially expressed genes (DEGs) identified among strains from different sources. Experimental verification confirmed that the differences in expression of bepC, secB, secDF, and ftsY played a key role in host-specific recognition. Furthermore, phylogenetic analysis based on transcriptomic data clearly reflected the taxonomic relationships among Bartonella species, indicating that their genetic evolution was primarily driven by host-related factors, a finding consistent with genome-based analysis.
Conclusions: Transcriptome data provides a powerful approach for clarifying species differentiation and evolutionary relationships within Bartonella spp., with potential applicability to other prokaryotic species. These findings provided critical insights for resolving taxonomic uncertainties and advancing systematic research.
-
Bartonella spp. are aerobic, fastidious intracellular parasites transmitted by arthropod vectors or through animal scratches (1). These pathogens exhibit broad host ranges and cause a spectrum of zoonotic diseases in humans, ranging from self-limiting infections to fatal outcomes (1). RNA sequencing (RNA-seq) has emerged as a powerful tool for studying the genetic evolution of species, particularly in eukaryotic phylogenetics (2). However, its application to phylogenetic and species differentiation studies in prokaryotes remains limited. Previous phylogenetic analyses of Bartonella spp. have relied on single-gene or multi-gene approaches (3-4), yet the taxonomic criteria employed in these studies remain poorly defined.
Eighty-seven Bartonella strains obtained from the Chinese Center for Disease Control and Prevention were analyzed (
Supplementary Table S1 ). Total RNA was extracted following cultivation and used for cDNA library construction. Paired-end sequencing was performed on the Illumina NovaSeq 6000 platform, followed by quality control, mRNA enrichment, and de novo transcriptome assembly. The assembled transcripts were functionally annotated against the Swiss-Prot, eggNOG, GO, and KEGG databases. Differentially expressed genes (DEGs) were identified using DESeq2 (|log2FoldChange|≥2, adjusted P<0.05) and subsequently subjected to GO and KEGG enrichment analyses. Single nucleotide polymorphisms (SNPs) in core genes were called from both transcriptome and genome data using Snippy (version 4.6.0, The University of Melbourne, Melbourne, Australia) and Gubbins (version 3.3.5, Wellcome Sanger Institute, Cambridge, UK), with B. grahamii (ATCC 700132) as the reference. Maximum likelihood (ML) and Bayesian inference (BI) phylogenetic trees were constructed using IQ-TREE (version 2.3.6, Center for Integrative Bioinformatics Vienna, Vienna, Austria) and MrBayes (version 3.2.7, Department of Biodiversity Informatics, Swedish Museum of Natural History, Stockholm, Sweden), respectively. DEGs were validated by quantitative polymerase chain reaction (qPCR) using gltA as the reference gene.After quality control, 2.18 billion clean reads were obtained. De novo assembly yielded a reference transcriptome comprising 51,639 transcripts, of which 34,069 unigenes were functionally annotated. Hierarchical clustering revealed divergent gene expression patterns that correlated with host origin (Figure 1A). Strains of canine and monkey origin formed distinct clusters characterized by upregulation of class I and II genes. In contrast, rodent-origin strains exhibited heterogeneous expression profiles. Notably, numerous DEGs were identified between different species and host orders, whereas far fewer were detected within the same species or host order (
Supplementary Tables S2 –S3 ). Species-level DEGs were enriched in broad metabolic and information-processing categories; however, host-order-level DEGs were strikingly enriched in functions related to host interaction and pathogenesis. Specifically, significant GO terms involved host cell components, the type IV secretion system (T4SS), and immune defense mechanisms. KEGG analysis further confirmed enrichment in key pathways, including bacterial secretion and prokaryotic defense systems. Given this enrichment in host-recognition-related secretion systems, we examined the expression of these genes in detail. Key genes encoding T4SS structural components, including virB2–4, were significantly upregulated in strains from the order Carnivora (Table 1). qPCR confirmed significant differential expression of bepC, secB, and secDF (P<0.05), with trends consistent with the RNA-seq data (Figure 1B–E,Supplementary Table S4 ).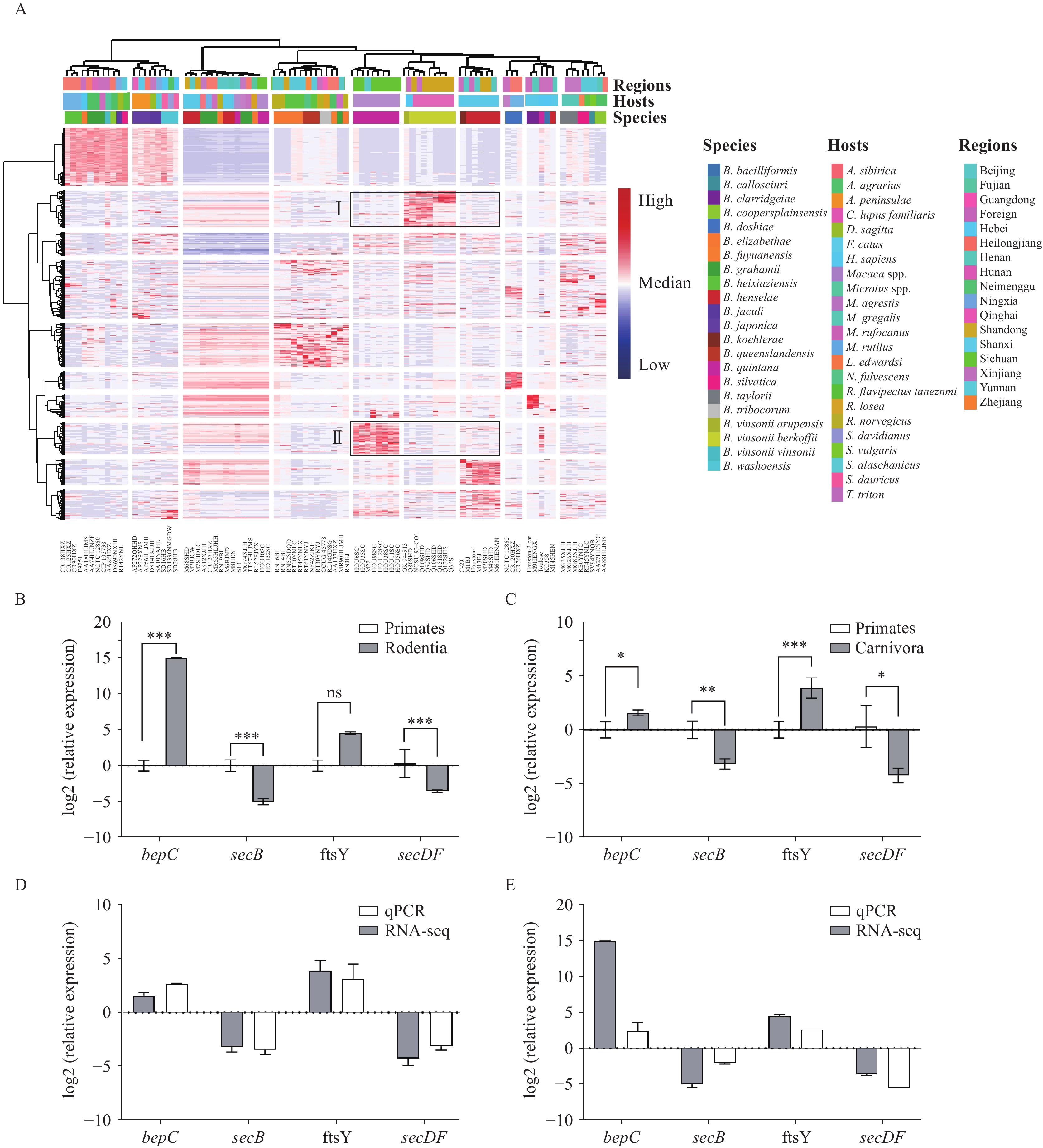 Figure 1.
Figure 1.Transcriptome analysis results. (A) Gene expression clustering heatmap of 87 Bartonella strains. (B) qPCR validation of selected DEGs from the “Primates vs. Rodentia” comparison. (C) qPCR validation of selected DEGs from the “Primates vs. Carnivora” comparison. (D) Comparison of qPCR and RNA-seq fold-change values for the “Primates vs. Rodentia” comparison. (E) Comparison of qPCR and RNA-seq fold-change values for the “Primates vs. Carnivora” comparison.
Abbreviation: ns=not significant; qPCR=quantitative polymerase chain reaction; DEG=differentially expressed genes.
* P<0.05, ** P<0.01, *** P<0.001.
Phylogenetic analyses were conducted independently using transcriptomic and genomic datasets. For the transcriptomic and genomic analyses, 19 and 31 reference genomes were incorporated, respectively, yielding 3,498 and 24,395 core gene SNPs. Despite the difference in reference genome numbers, comparative analysis confirmed that this did not affect reconstruction reliability (Figure 2A–B). Transcriptome trees were fully resolved and exhibited strong nodal support (ML≥75%, BI≥85%). All species-level branches displayed clear species-specific clustering, revealing closely related pairs such as B. koehlerae–B. henselae and B. doshiae–B. bacilliformis. The genome-based tree also demonstrated clear species clustering with even higher support values (ML=100%, BI=100%). While interspecific relationships were largely congruent between tree types, discrepancies were observed; notably, B. bacilliformis clustered more closely with B. clarridgeiae in the genome tree. Despite these minor topological differences, species-level clade structures were highly consistent across datasets. Both ML trees exhibited clear host-associated clustering at the order level (Rodentia, Primates, and Carnivora) and at finer host-species resolution. For example, dog-associated B. vinsonii subsp. berkhoffii, cat-associated B. koehlerae and B. henselae, and monkey-associated B. quintana formed distinct clusters. In contrast, geographical distribution had limited influence on clustering patterns. Apart from some strains from Sichuan and Shandong provinces forming clusters, the remaining strains showed a mosaic distribution without strong geographical signal.
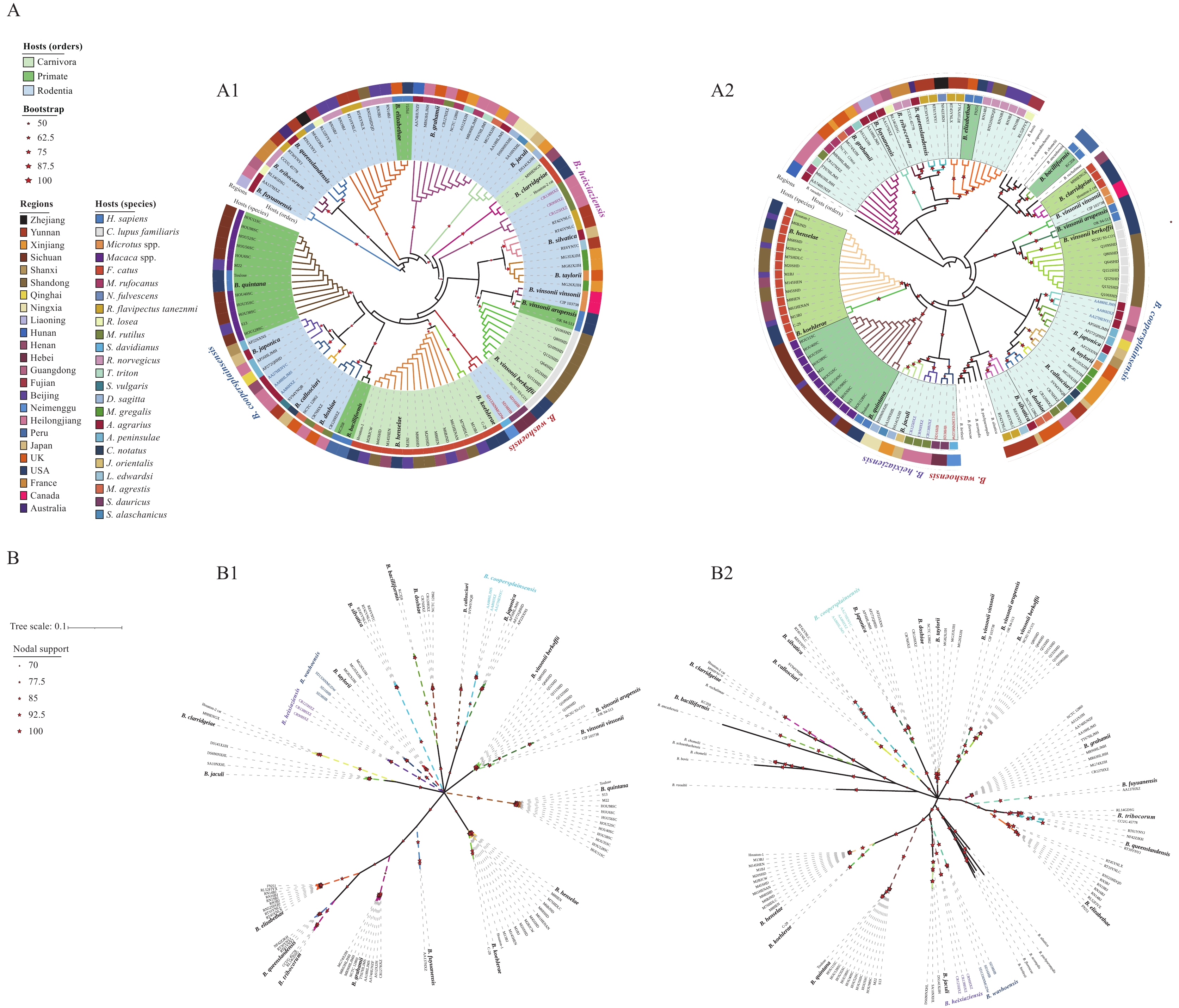 Figure 2.
Figure 2.Phylogenetic trees based on core-gene SNPs. (A) ML trees constructed from transcriptomic (A1) and genomic (A2) data; (B) BI trees constructed from transcriptomic (B1) and genomic (B2) data.
Note: Branch colors indicate Bartonella species. Reference genomes are shown in bold; species lacking reference genomes are labeled on an outer ring with matching colors.
Abbreviation: SNP=Single nucleotide polymorphism; ML=Maximum likelihood; BI=Bayesian inference.
-
Consistent with the role of gene regulation in adaptation, hierarchical clustering revealed that Bartonella gene expression was conserved within species but variable between species (5). Furthermore, transcriptome composition differed significantly across species and host origin. Host specificity has been linked to the ability of pathogens to inject effector proteins into host cells via specialized secretion systems (6). Previous studies have demonstrated that Bartonella spp. rely primarily on the VirB/VirD4 T4SS to transport effector proteins into host cells, thereby modulating immune responses and disrupting cellular structures to promote colonization (7). In the present study, differential expression of virB and bepC was confirmed, and several novel host-associated DEGs were identified, including tatC, tatA, and ftsY. Given their implicated roles in virulence and adaptation in other pathogens, these genes may contribute to host-specific recognition in Bartonella spp. (8).
Traditional phylogenetic analyses have relied on mitochondrial and a limited number of nuclear gene markers, which are susceptible to stochastic errors (9). Advances in high-throughput RNA-seq now enable the application of comprehensive transcriptomic data to phylogenetic studies. In the present analysis, phylogenetic reconstruction based on core-gene SNPs from both transcriptomic and genomic data revealed highly congruent tree topologies, validating the utility of transcriptomes for resolving taxonomic relationships within Bartonella spp. These analyses further revealed significant clustering of strains according to host origin, indicating that host association acted as a primary driver of species differentiation in Bartonella spp. In contrast to previous studies which had emphasized the role of geographic distribution in the genetic differentiation of Bartonella, our findings suggested that its influence was comparatively limited (4). This pattern could be attributed to increased cross-regional movement of host animals driven by modern socioeconomic development, which likely attenuated the impact of geographic barriers on the genetic evolution of Bartonella spp.
This study has several limitations. First, the limited number of strains representing certain Bartonella species, host types, and geographic regions may affect the generalizability of these findings. Future studies should expand the strain collection to determine whether expression profiles exhibit stronger host-specific or geographic clustering patterns. Second, the molecular mechanisms underlying the identified host-associated genes remain uncharacterized. Subsequent investigations should employ transcriptional regulation experiments to define the functional roles of these genes in host adaptation.
In conclusion, the transcriptome of Bartonella spp. varied systematically by species and host origin, supporting a model of host-driven speciation and evolution. This study demonstrates the utility of transcriptomics for elucidating evolutionary relationships in Bartonella spp. and potentially in other prokaryotes.
HTML
| Citation: |

|