
 Download:
Download:




-
Human adenoviruses (HAdVs) are non-enveloped, double-stranded DNA viruses (genome size 34–36 kb) in the genus Mastadenovirus (family Adenoviridae), classified into 7 species (A–G) with 116 recognized types (http://hadvwg.gmu.edu/) (1). Among these, HAdV-3, HAdV-7, HAdV-55, HAdV-4, and species C (HAdV-C) are the major epidemiologically significant causes of respiratory infections (2–6).
HAdV-21 (species B), first identified in Saudi Arabia in 1956, is increasingly recognized as a significant pathogen in military and civilian populations across multiple countries (7). Although a meta-analysis of Chinese surveillance data (2009–2021) revealed a low prevalence of HAdV-21 (0.87%), recent data from 14 sentinel surveillance sites across 11 provincial-level administrative divisions (PLADs) (2023–2024) indicated a marked increase: the proportion of HAdV-21 infections rose from 1.44% (2/139) in 2023 to 8.97% (21/234) in 2024 (Fisher’s exact test: P=0.003) (unpublished data) (6). However, molecular epidemiological data on HAdV-21 in China remain limited, with only 3 isolates reported before 2023 (8–9).
To investigate the potential genetic correlations among the increasingly detected HAdV-21, this study performed whole-genome sequencing (WGS) of 23 HAdV-21 strains isolated from acute respiratory infections across 7 surveillance PLADs (2023–2024) and conducted phylogenetic, genetic variation, and recombination analyses by integrating these data with previously reported HAdV-21 genomes obtained from GenBank.
-
Between 2023 and 2024, 23 HAdV-21 strains were isolated from 7 Chinese sentinel surveillance PLADs (Gansu, Jilin, Ningxia, Liaoning, Guangdong, Chongqing, and Beijing) and confirmed by real-time polymerase chain reaction and amplification/analysis of 3 genes (penton base, hexon, and fiber). The corresponding 23 clinical cases ranged from 5 months to 68 years (median: 3 years). Among these cases, 14 (60.87%) required hospitalization. Ten cases presented with upper respiratory tract infections, while the remaining 13 had lower respiratory tract infections, including 10 with pneumonia. Co-infections occurred in 13 cases, most commonly with Haemophilus influenzae (5/13). One fatal case (Ningxia2024-669) of acute respiratory distress syndrome exhibited Mycoplasma pneumoniae co-infection (Table 1).
Strain PLADs Gender Age Case type Year of collection Co-infection pathogens Clinical diagnosis Subtype Jilin2023-296 Jilin Female 3 years Inpatient 2023/5/21 / Bronchopneumonia HAdV-21a Jilin2023-595 Jilin Female 3 years Inpatient 2023/6/19 / Upper respiratory infection HAdV-21a Ningxia2024-533 Ningxia Male 3 years Outpatient 2024/2/10 SARS-CoV-2 Fever HAdV-21a Ningxia2024-595 Ningxia Female 5 months Inpatient 2024/3/4 Hi Severe lung infection/Lung consolidation HAdV-21a Ningxia2024-608 Ningxia Female 5 years Outpatient 2024/3/1 MP Acute tonsillitis HAdV-21a Ningxia2024-618 Ningxia Male 13 years Outpatient 2024/3/5 Hi Upper respiratory infection HAdV-21a Ningxia2024-669* Ningxia Male 9 months Inpatient 2024/3/14 MP Severe lung infection/ARDS HAdV-21a Ningxia2024-671 Ningxia Female 5 years Inpatient 2024/3/18 / Severe lung infection/Lung consolidation HAdV-21a Ningxia2024-725 Ningxia Male 13 years Outpatient 2024/3/27 MP/Flu B Respiratory infection HAdV-21a Ningxia2024-747 Ningxia Male 6 years Inpatient 2024/4/1 MP Lung infection/Acute suppurative tonsillitis HAdV-21a Ningxia2024-754 Ningxia Female 3 years Outpatient 2024/3/31 Hi Acute bronchitis HAdV-21a Ningxia2024-868 Ningxia Female 4 years Outpatient 2024/4/29 SP/Hi Respiratory infection HAdV-21a Ningxia2024-907 Ningxia Female 7 months Outpatient 2024/5/8 Hi Respiratory infection HAdV-21a Ningxia2024-944 Ningxia Male 2 years Outpatient 2024/5/21 SP Fever HAdV-21a Beijing2024-330 Beijing Female 3 years Inpatient 2024/3/28 HRV Bronchopneumonia HAdV-21a Beijing2024-364 Beijing Male 1 year Inpatient 2024/5/6 / Pneumonia HAdV-21a Beijing2024-378 Beijing Male 2 years Inpatient 2024/5/21 Flu B Fever HAdV-21a Shenzhen2024-077 Guangdong Male 8 months Inpatient 2024/4/27 / Pneumonia HAdV-21a Chongqing2024-069 Chongqing Female 2 years Inpatient 2024/4/2 / Febrile convulsions/Bronchitis HAdV-21a Shenyang2024-163 Liaoning Male 57 years Outpatient 2024/3/12 / Respiratory infection HAdV-21a Gansu2024-215 Gansu Male 68 years Inpatient 2024/2/17 / Community-acquired pneumonia HAdV-21a Ningxia2024-674 Ningxia Male 1 year Inpatient 2024/3/18 / Severe lung infection HAdV-21b Shenzhen2024-201 Guangdong Male 3 years Inpatient 2024/6/11 / Acute laryngotracheobronchitis HAdV-21b * Indicates fatalities.
Note: / means no co-infecting pathogens detected.
Abbreviation: ARDS=acute respiratory distress syndrome; PLAD=provincial-level administrative division; Hi=Haemophilus influenzae; MP=Mycoplasma pneumoniae; Flu B=Influenza B virus; SP=Streptococcus pneumoniae.Table 1. Detailed information on the 23 HAdV-21 strains analyzed in this study.
Viral DNA from the 23 strains was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). WGS was performed by iGeneTech Biotechnology Co., Ltd. using probe-based hybrid capture combined with next-generation sequencing. All strains achieved 100% genome coverage, each yielding >1 Gb of data at >8,000× depth. Suboptimal regions were validated by Sanger sequencing, and two strains were randomly resequenced for quality assurance. Genome annotation was performed in Geneious Prime (version 2023.2.2, Biomatters Ltd., Auckland, New Zealand) using the HAdV-21 prototype strain (AY601633) as a reference.
Fifty HAdV-21 genomes from 6 countries (1956–2019), including 3 from China (2019) and 47 from other countries (1956–2018), were retrieved from GenBank (
Supplementary Table S1 ). Integration of the 23 newly sequenced strains yielded a comprehensive database of 73 WGSs for phylogenetic and genetic variation analyses.Sequences were aligned using ClustalW, and sequence similarity was assessed using BioEdit. A maximum likelihood phylogenetic tree was constructed using MEGA Version 7.0, with bootstrap support values (>80%) indicated at the tree nodes. Genetic mutations were identified using Snipit (https://github.com/aineniamh/snipit), focusing on variations with frequencies >80%. Recombination was analyzed using SimPlot (window size: 1,000 bp; step size: 100 bp) and Recombination Detection Program v4 (RDP4).
-
All 23 HAdV-21 strains were fully assembled (genome sizes: 35,364–35,393 bp; GC: approximately 51.2%), consistent with previous reports (8–9). Annotation using the HAdV-21 prototype strain as a reference identified 48 conserved protein-coding regions (
Supplementary Table S2 ). Sequence identity analysis among the 23 HAdV-21 strains was ≥99.7%. Strains with identical sequences were detected across different PLADs and years, e.g., the 2023 Jilin strains (Jilin2023-296 and Jilin2023-595) matched 2024 strains from 5 PLADs (Beijing2024-364, Gansu2024-215, Ningxia2024-907, Shenyang2024-163, and Shenzhen2024-077). Identical sequences were also detected within the same PLAD, such as Ningxia2024-595 and Ningxia2024-725.The strains shared 98.8% nucleotide identity with the HAdV-21 prototype strain. Analysis of 12 functional domains, including major capsid proteins (penton base, hexon, fiber), core proteins (pV, pVII, pTP, pIVa2), minor proteins (pIX, pIIIa, pVI, pVIII), and the non-structural protein pX, showed relatively high similarity (nt: 98.4%–99.8%, aa: 97.9%–100.0%) across 11 regions, with the penton base showing lower conservation (nt: 96.3%, aa: 95.7%–95.9%).
-
To determine the genetic relationships between the 23 HAdV-21 strains (2023–2024) and previously circulating Chinese and globally prevalent strains, a phylogenetic analysis was performed using the constructed WGS dataset (Figure 1). The results showed that the 73 HAdV-21 strains were categorized into 3 distinct evolutionary lineages: HAdV-21a, HAdV-21b, and HAdV-21p (bootstrap support >80%). HAdV-21a and HAdV-21b (HAdV-21a/b) exhibited closer phylogenetic relationships to each other (genetic distance=0.001) than to HAdV-21p (genetic distance=0.007 for both subtypes), indicating a shared ancestry for HAdV-21a/b.
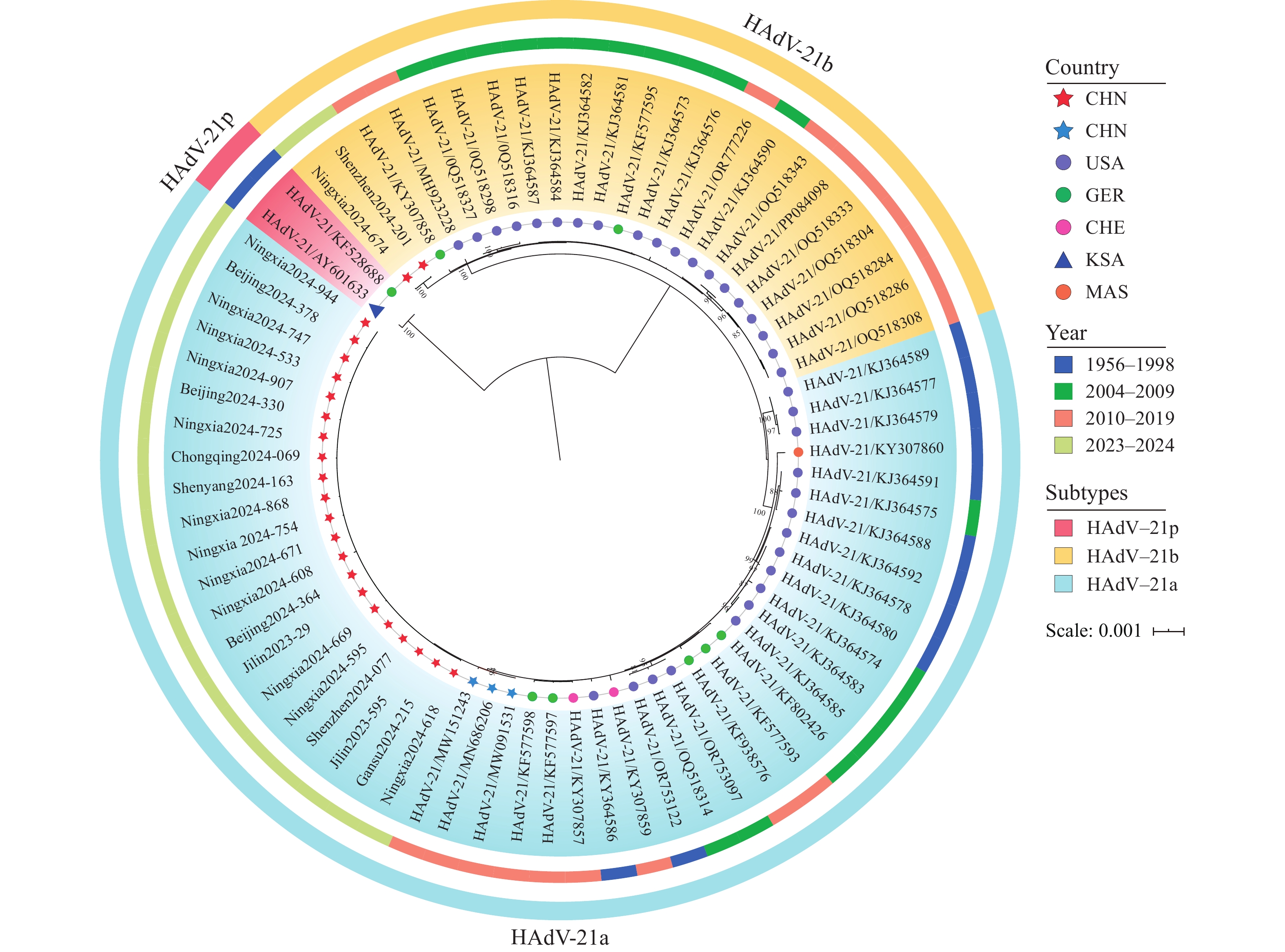 Figure 1.
Figure 1.Maximum-likelihood phylogenetic trees of HAdV-21 were constructed from a dataset of 73 strains (23 from this study and 50 from the GenBank database). Trees derived from nine individual gene fragments of HAdV-21 are presented in panels A-I, and the tree based on the WGS is presented in panel J. The HAdV-21 and HAdV-3 prototype strains are marked in red and dark blue, respectively, across all panels
Abbreviation: WGS=whole-genome sequencing; HAdV-21=human adenovirus type 21.Further analyses revealed distinct temporal and geographical distribution patterns among the 3 HAdV-21 subtypes. HAdV-21p primarily comprised a prototype strain from Saudi Arabia and a historical strain from Germany (1950s). Conversely, HAdV-21a comprised 21 strains from 7 Chinese PLADs (2023–2024), 3 Chinese strains from 2 PLADs (2019), and 24 strains from the United States, Malaysia, Germany, and Switzerland (1956–2016). HAdV-21b included 2 strains from 2 Chinese PLADs (2024) and 21 strains from the United States and Germany (2005–2018). All 3 subtypes demonstrated minimal intra-subtype genetic variation (distance: 0.00014–0.00031). HAdV-21a/b demonstrated high genetic consistency between 2023–2024 Chinese strains and both the 2019 Chinese strains (distance: 0.00007) and global strains (distance <0.00040), strongly suggesting close genetic relationships. No significant correlation was observed between clinical severity and 2 subtypes (HAdV-21a and HAdV-21b).
-
Genetic variation analysis of HAdV-21a/b was performed using WGS with the HAdV-21p prototype strain as a reference. The results showed that the 2 subtypes shared 352 specific nucleotide variants, including 64 insertions and 49 deletions, with HAdV-21a exhibiting 27 unique variants (10 deletions) and HAdV-21b displaying 36 unique variants (3 insertions and 18 deletions). The variants were distributed across the genome, with insertions and deletions concentrated in the L2 (46.90%) and E3 (23.01%) regions, respectively. Non-coding region variants were also detected in the Chinese HAdV-21a (4 sites, 1 insertion) and HAdV-21b (7 sites).
Amino acid variation analysis across the 12 functional domains identified 70 shared HAdV-21a/b substitutions in 11 proteins, excluding pX (Figure 2). 9 of 10 shared hexon gene variants were localized to hypervariable regions (HVRs), with HVR7 exhibiting the highest mutation frequency (4 sites). In the fiber gene, 2 of 3 variants occurred in the shaft region and one in the knob region. The penton base gene exhibited 23 variations, primarily clustered within the RGD loop, including 15 amino acid deletions (313TEAAKAAAIAKANIV327) and 2 insertions (362EE363). Subtype-specific mutations were also detected, including pIVa2 (HAdV-21b: L196F), pTP (HAdV-21a: D45N), and penton base (HAdV-21b: E213D and S349F).
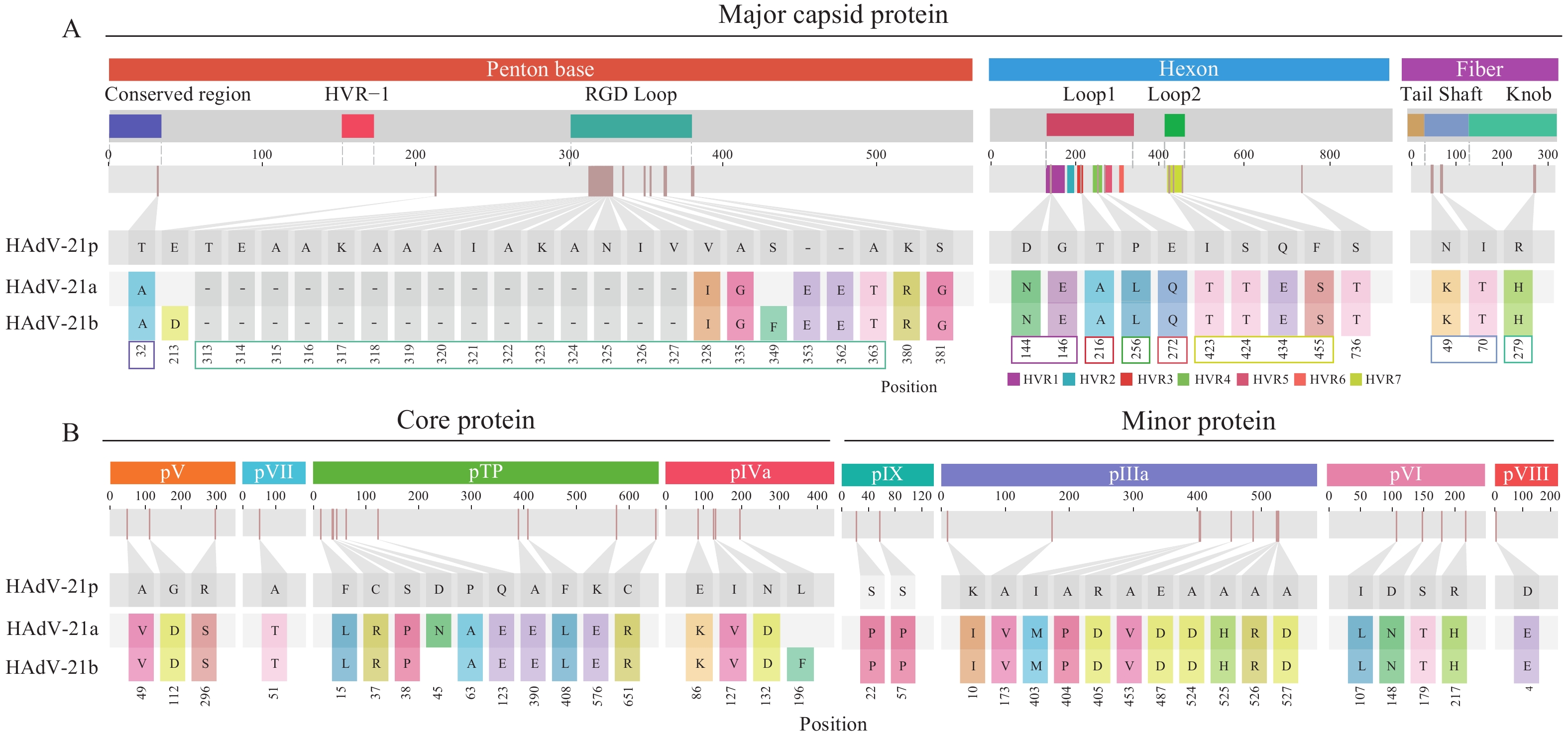 Figure 2.
Figure 2.Amino acid variations in 11 key proteins of (A) HAdV-21a and (B) HAdV-21b subtypes compared to the prototype HAdV-21 strain (designated HAdV-21p).
Abbreviation: HAdV-21=human adenovirus type 21. -
To assess recombination, phylogenetic analysis was performed with the HAdV-21 strains and 18 species B prototype strains (
Supplementary Table S1 ) using 9 consecutive genomic fragments (nt1–7,000; nt7,001–13,877; penton base; nt15,564–18,453; hexon; nt21,304–26,000; nt26,001–31,405; fiber; nt32,378–end) and WGS (Supplementary Figure S1 ). The results demonstrated that subtype classification using the nt32,378–end fragment matched the WGS-based classification, whereas the other 8 fragments failed to distinguish HAdV-21a/b. In the nt32,378–end region, HAdV-21a/b exhibited closer phylogenetic relationships with the HAdV-3 prototype strain, suggesting potential recombination in the E4 region. SimPlot and RDP4 analyses (supported by four algorithms) further confirmed identical recombination patterns in both subtypes, with a breakpoint at nt32,843 in the E4 gene (Figure 3).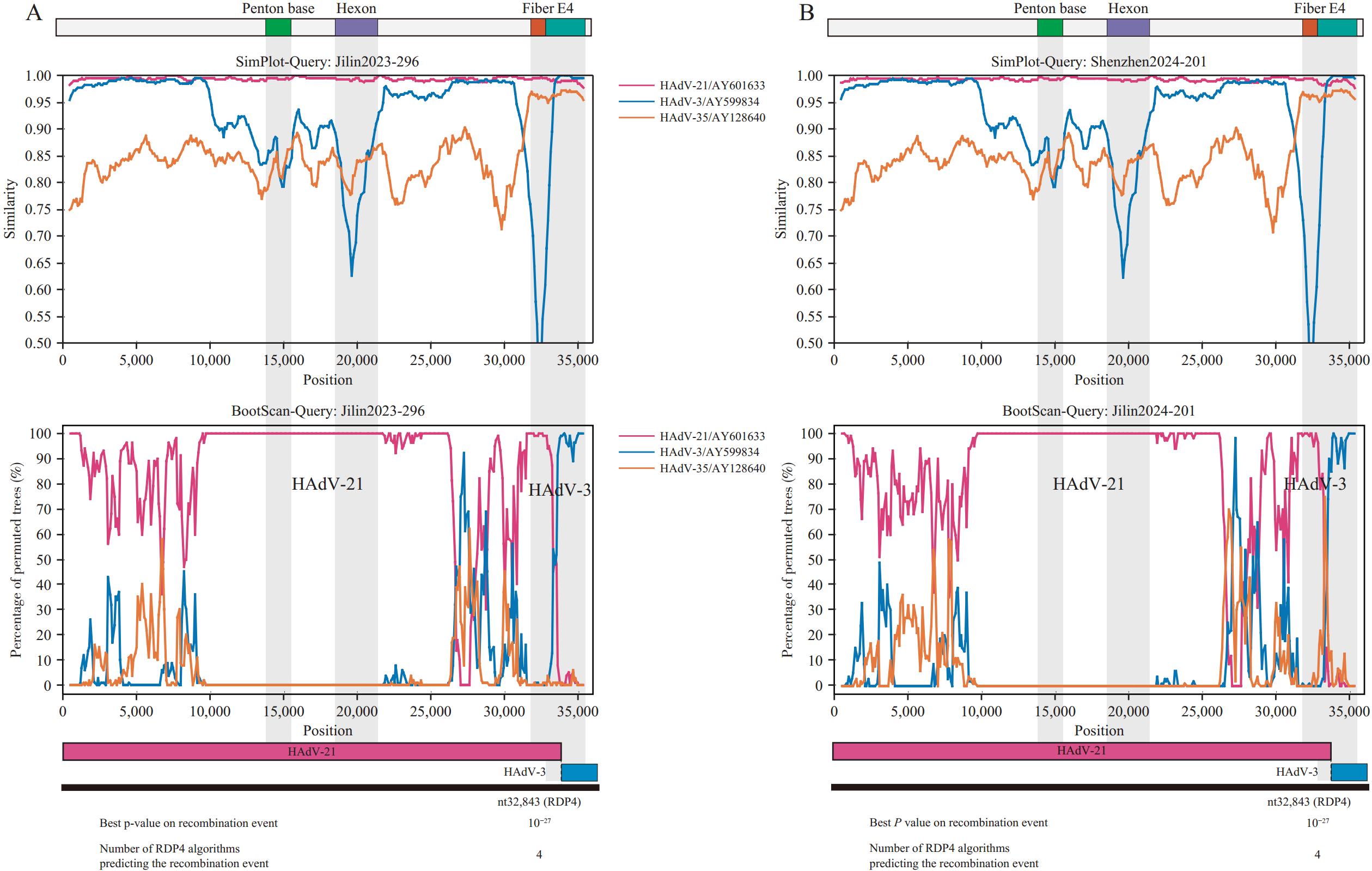 Figure 3.
Figure 3.Recombinant analysis of (A) HAdV-21a and (B) HAdV-21b strains.
Note: Jilin2023-296 and Shenzhen2024-201 were used as representative HAdV-21a and HAdV-21b strains, respectively.
Abbreviation: HAdV-21=human adenovirus type 21.
-
This study analyzed 23 HAdV-21 strains (2023–2024) isolated from patients with acute respiratory infections. Among these, 56.5% (13/23) had lower respiratory tract infections, including 9 pneumonia cases and 1 fatal infant case. To elucidate the genetic basis of these emerging strains, this study performed a comprehensive genomic characterization.
Phylogenetic analyses identified viral genetic subtypes and spatiotemporal transmission patterns. Globally, HAdV-21 strains are classified into 3 subtypes: HAdV-21a, HAdV-21b, and HAdV-21p. HAdV-21p, comprising historical strains from the 1950s, is no longer detected. Conversely, HAdV-21a (1956–2024, 5 countries) and HAdV-21b (2005–2024, 3 countries) demonstrated extensive spatiotemporal distribution, reflecting stable epidemic trends and highlighting these strains as the dominant circulating subtypes in multiple countries, including China. Furthermore, all subtypes exhibited minimal intra-subtype genetic variation (distance <0.00031), suggesting genomic stability during prolonged circulation, consistent with observations in other HAdV types, such as HAdV-4 and HAdV-55 (10-11). This stability may result from the conserved replication mechanism of HAdV or selective equilibrium under host immune pressure.
Notably, Chinese HAdV-21a/b strains (2023–2024) exhibited extremely high genetic homology and consistency with 2019 Chinese strains (distance: 0.00007) and historical global strains (distance: <0.00040), indicating that the currently circulating HAdV-21 is not a novel variant but a continuous transmission of genetically related strains. Enhanced respiratory infection surveillance following coronavirus disease 2019 (COVID-19) likely contributed to increased detection rates in recent years. Although HAdV-21 demonstrates relatively lower virulence and infectivity than other common types (e.g., HAdV-3, -7, -4, -55), it is associated with diverse clinical manifestations, including severe pneumonia, acute respiratory distress syndrome, acute flaccid paralysis, myocarditis, and fatal outcomes across age groups, necessitating urgent research on its pathogenic characteristics (8–9,12–14).
Phylogenetic and recombination analyses confirmed a common ancestor for HAdV-21a/b. Despite high overall genome conservation, sequence variation analysis identified both shared and subtype-specific mutations in HAdV-21a/b, representing key molecular markers for subtype differentiation and indicating continuous adaptive evolutionary pressure. Mutations in the hexon gene are predominantly localized in HVRs containing critical neutralizing antibody epitopes, suggesting that HVRs are primary adaptive targets for immune evasion. Consistent with previous studies, HAdV-21a/b exhibited 15 amino acid deletions and 2 insertions in the RGD loop of the penton base gene compared to the prototype strain. This shortened RGD loop closely resembles those of HAdV-3 and HAdV-7, which are associated with severe diseases (15). Because the RGD loop is a core functional domain mediating viral cell entry, its alteration may directly affect viral pathogenicity and infection efficiency; therefore, the functional consequences of the penton base warrant further investigation.
Recombination analysis revealed that HAdV-21a/b acquired an HAdV-3-derived fragment in the E4 region (breakpoint: nt32,843). As E4 regulates viral DNA replication, transcript splicing, and late gene expression, this recombination likely enhances viral stability and replication efficiency through functional complementation, thereby conferring transmission advantages. Genetic recombination drives HAdV evolution, where adaptive recombination patterns establish stable transmission, as exemplified by HAdV-21a and HAdV-21b persisting in circulation for approximately 70 and 20 years, respectively.
The findings in this report are subject to at least three limitations. First, HAdV-21 strains were collected from only 7 Chinese PLADs, limiting the generalizability of the findings; broader sampling in future work would improve representativeness. Second, the impact of HAdV-21 genomic differences on viral infection phenotypes remains unclear, warranting further investigation. Finally, although this study identified a significant HAdV-3 fragment-containing recombination event, functional validation was lacking, necessitating additional experiments to clarify its implications for viral pathogenicity and transmission.
In conclusion, this study identified the co-circulation of HAdV-21a and HAdV-21b in China and characterized their genomic features. Given HAdV-21’s increasing prevalence, persistence of similar viruses across multiple PLADs, and potential association with severe clinical outcomes, enhanced surveillance is required to quantify disease burden in China and provide targeted prevention and control strategies.
-
The study used only viral strains and did not involve specimen collection. We sincerely thank the seven sentinel surveillance PLADs for providing the viral strains and associated data.
HTML
Genomic Characterization
Phylogenetic Analysis
Genetic Variation
Genetic Recombination
| Citation: |

|