
 Download:
Download:




-
Introduction: Suidae-associated zoonotic viruses represent a significant global public health threat through cross-species transmission events. Current research remains limited to localized outbreak investigations and lacks comprehensive, systematic global analysis.
Methods: We collected human–Suidae virus data from the National Center for Biotechnology Information (NCBI) Virus Database, integrating viral characteristics, host information, and environmental and anthropogenic factors. Boosted Regression Trees (BRT) models were employed to evaluate cross-species transmission risk and identify key predictive factors.
Results: A total of 43 human-Suidae zoonotic viruses reported durng 1882–2022 were evaluated. The Boosted Regression Trees (BRT) model achieved area under the curve (AUC) values of 0.924 (training) and 0.804 (testing). Host–human phylogenetic distance and viral genome size emerged as the primary predictors. Porcine circovirus 3 (PCV3) demonstrated the highest predicted risk (>0.9).
Conclusions: This study establishes a data-driven framework for assessing cross-species transmission risk, supporting early warning systems and targeted prevention strategies. The findings underscore the critical importance of One Health approaches and recommend enhanced surveillance and biosecurity measures for high-risk viruses such as PCV3.
-
Zoonotic diseases represent a major threat to global public health, accounting for nearly 60% of recognized human diseases and approximately 75% of emerging infectious diseases in recent decades (1). Among animal reservoirs, Suidae (pig family) are of particular concern due to their widespread distribution, intensive farming practices, and close contact with humans, which establish them as critical intermediate hosts facilitating viral spillover events (2).
Although substantial progress has been achieved in understanding transmission mechanisms of individual viruses such as influenza and Japanese encephalitis, most existing studies remain fragmented — focusing on single pathogens, geographically restricted regions, or lacking comprehensive integration across viral, host, and environmental dimensions (3-4). Consequently, the key drivers and spatiotemporal dynamics of zoonotic spillovers remain inadequately quantified.
To address these knowledge gaps, this study systematically analyzes globally reported human–Suidae zoonotic viruses using the National Center for Biotechnology Information (NCBI) Virus Database. By integrating viral genomic characteristics, host phylogenetic relationships, and environmental data, we developed a predictive model to identify the relative importance of risk factors and characterize high-risk viral species. These findings aim to provide an evidence-based framework for early warning systems and targeted interventions under the One Health approach, with particular relevance for major pork-producing countries such as China and the United States.
We retrieved spatiotemporal data on human-Suidae viruses and their hosts from the NCBI Virus Database (last accessed July 2025), including the 2025 data update, which verified that the initial sampling dates for all identified viruses remained consistent, defined cross-species transmission (CST) events, and integrated external datasets to explore potential influencing factors and their relative importance in shaping these transmission patterns (
Supplementary Figure S1 ).After removing duplicates and incomplete entries, 813,809 entries were retained, from which we identified 43 viruses infecting both humans and Suidae, yielding 188,302 records. These zoonotic viruses were standardized using the International Committee on Taxonomy of Viruses (ICTV) Master Species List (2022.v1). Viral attributes were annotated using the ViralZone database, including genome type, strandedness, segmentation, replication site, envelope presence, and genome size. Host taxonomy was verified through the Animal Diversity Web and NCBI Taxonomy, with evolutionary divergence times between host families and humans or Suidae estimated using TimeTree as a proxy for genetic distance. Environmental and anthropogenic factors were compiled from WorldClim and FAOSTAT databases, including long-term climate indicators (temperature and precipitation), artificial surface area (representing human environmental modification such as roads, buildings, and other impervious infrastructure), urban/rural population densities, and World Bank income classifications. Cross-species transmission events were defined following the host-association framework established by Olival et al. (5), whereby viruses empirically detected in both humans and Suidae were classified as zoonotic, reflecting their biological capacity to cross host barriers regardless of confirmed transmission directionality.
Predictive modeling was performed to evaluate CST risk by integrating viral, host, and environmental attributes. We tested logistic regression and machine learning models, selecting the Boosted Regression Tree (BRT) as the primary approach (Table1) (6). Hyperparameters were optimized through grid search and 10-fold cross-validation. The final model employed a learning rate of 0.01, tree complexity (interaction depth) of 3, and Bernoulli deviance loss function appropriate for binary classification. We allowed up to 10,000 trees with early stopping based on minimum cross-validation deviance. A bag fraction of 0.5 was applied to introduce stochasticity and reduce overfitting. Model performance was monitored across folds, and variable importance was assessed based on relative influence of each predictor across all fitted trees. Model performance was evaluated using a stratified 7:3 training–testing split, with metrics including sensitivity, specificity, accuracy, balanced accuracy, and the area under the receiver operating characteristic curve (AUC). Variable importance was quantified by relative influence across fitted trees, with predictors considered statistically significant if P< 0.05 and 95% confidence intervals excluded zero. All statistical analyses were performed using R statistical software (version 4.1.0, The R Foundation for Statistical Computing, Vienna, Austria).
Among the evaluated models, the BRT demonstrated superior performance, achieving an AUC of 0.924 for the training dataset and 0.804 for the test dataset (Figure 1). These evaluations were based on 43 human–Suidae viruses reported during 1882–2022, indicating robust predictive capability with minimal overfitting, and confirming the model’s suitability for assessing CST risk among human-Suidae viruses.
Model Sensitivity Specificity Accuracy Balanced accuracy AUC LR 0.681 0.717 0.716 0.699 0.699 BRT 0.653 0.807 0.803 0.730 0.804 DT 0.702 0.744 0.743 0.723 0.723 RF 0.319 0.960 0.946 0.640 0.640 SVM 0.722 0.747 0.747 0.735 0.735 Abbreviation: LR=Logistic Regression, BRT=Boosted Regression Trees, DT=Decision Tree, RF=Random Forest, SVM=Support Vector Machine, AUC=area under the curve. Table 1. Comparison of performance metrics across multiple methods for predicting cross-species transmission risk.
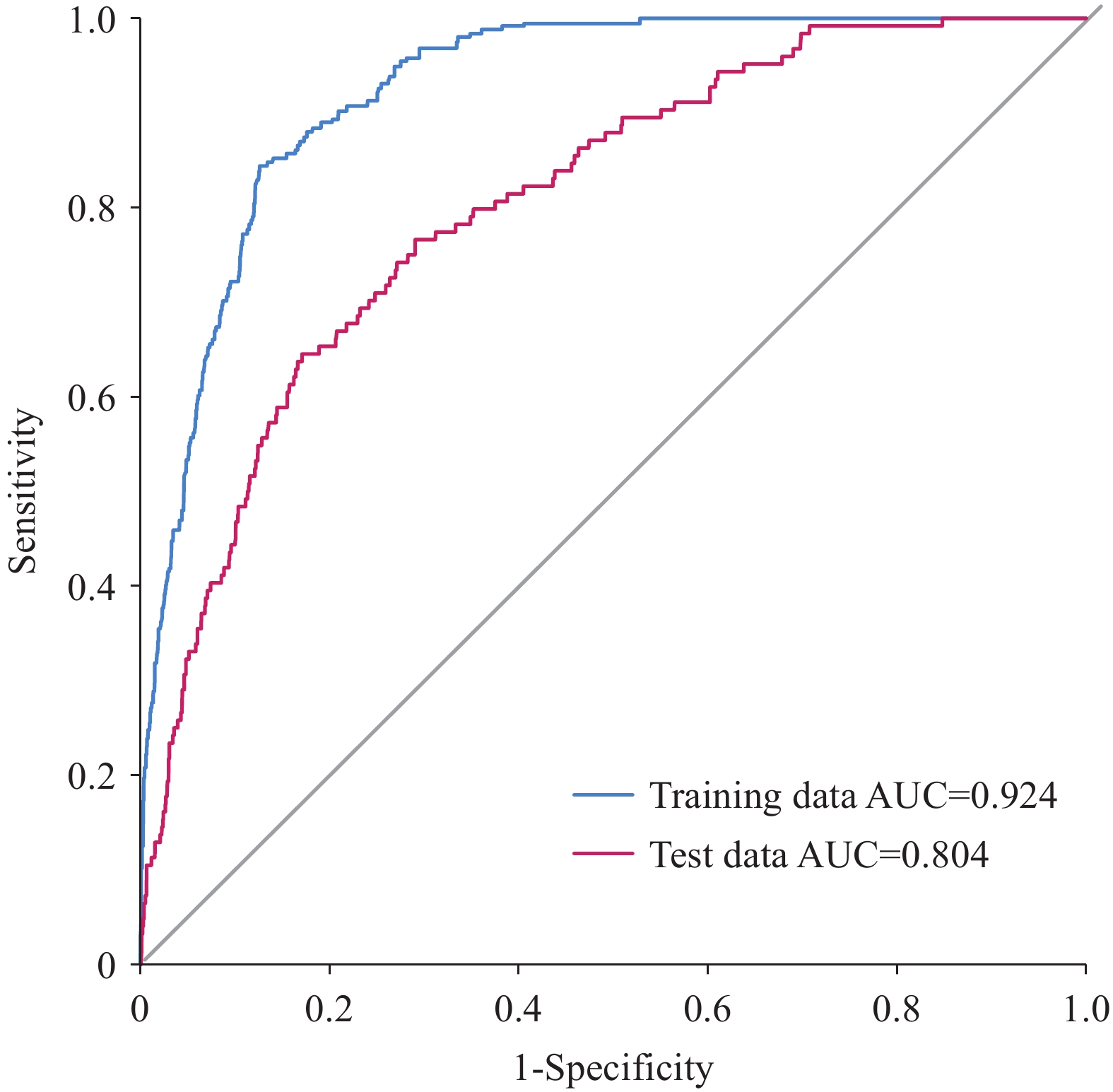 Figure 1.
Figure 1.ROC curves for training and test datasets using the BRT model.
Abbreviation: ROC=receiver operating characteristic; BRT=Boosted Regression Tree; AUC=area under the curve.The BRT analysis revealed eleven predictors with measurable contributions, with the top five demonstrating the greatest influence: host–human divergence time (28.50%), viral genome size (18.55%), rural human population density (11.29%), annual precipitation (9.72%), and artificial surface coverage (9.65%). These findings emphasize the critical importance of host phylogenetic proximity and viral genomic characteristics, alongside environmental and anthropogenic factors, in determining zoonotic transmission risk (Figure 2).
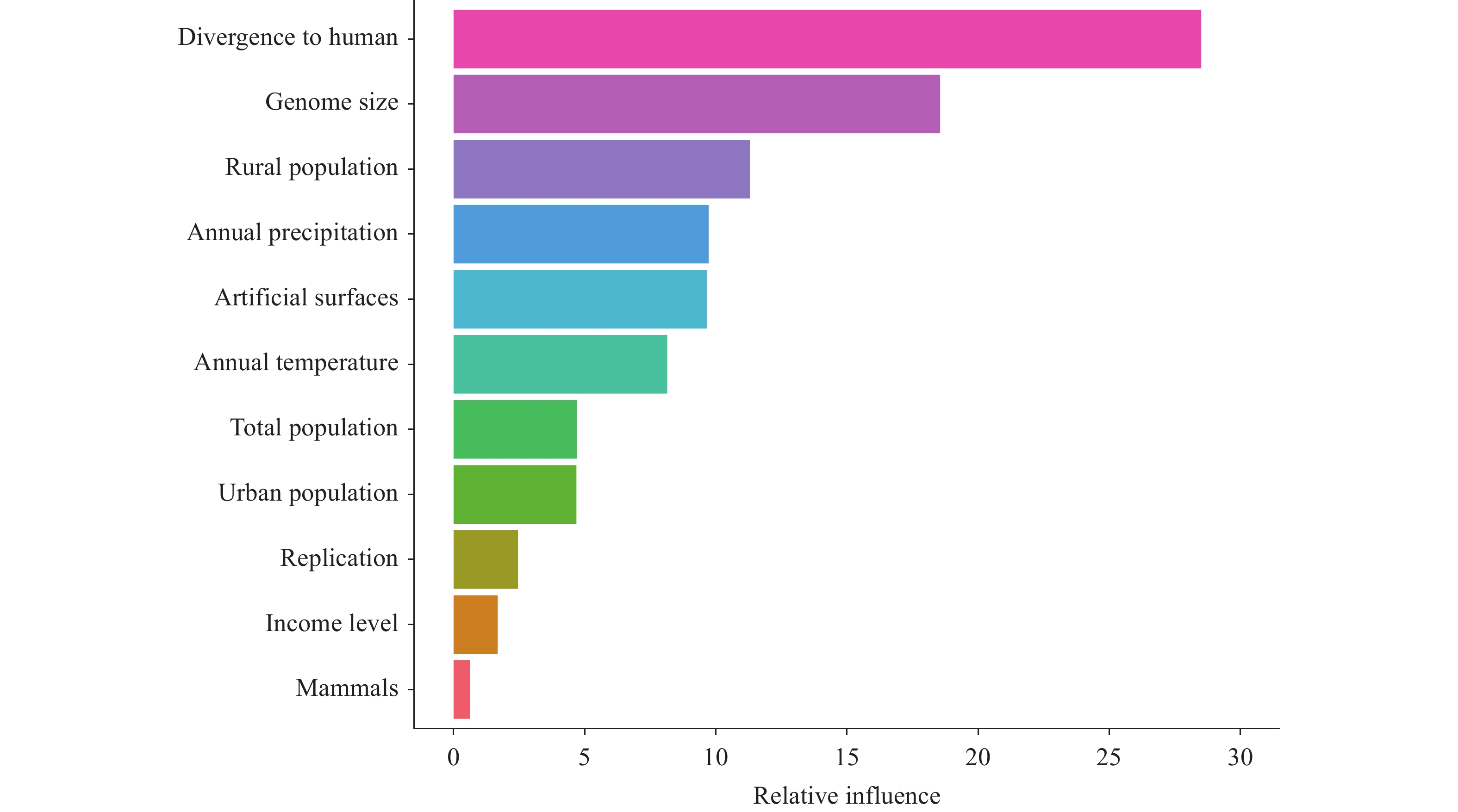 Figure 2.
Figure 2.Feature importance ranking in the BRT model.
Abbreviation: BRT=Boosted Regression Tree.Specifically, viruses originating from hosts with closer genetic relationships to humans demonstrated higher propensity for crossing species barriers, while viruses with larger genomes exhibited enhanced adaptive capacity for novel host environments. Rural population density and precipitation patterns reflected increased opportunities for human–animal interactions and favorable conditions for viral persistence, while land-use modifications indicated ecological disruptions that promote spillover events.
To extend the model’s application to Suidae viruses not yet documented in humans, we evaluated six common livestock viruses with the highest number of Suidae host records from the NCBI Virus database: Porcine reproductive and respiratory syndrome virus, African swine fever virus, Pestivirus C (classical swine fever virus), Foot-and-mouth disease virus, Porcine epidemic diarrhea virus, and Porcine circovirus 3.
Notably, Porcine circovirus 3 exhibited the highest predicted transmission risk, with probabilities exceeding 0.9 (Table 2). A predicted risk value >0.9 indicates that samples within this interval were assigned a probability exceeding 90% for cross-species transmission, reflecting high model confidence that these instances are associated with elevated spillover risk.
Predicted risk
level (%)Event
Probabilities(%)No. of
samples95% CI 0–15 0.5 1,796 (0.002, 0.009) 15–30 1.4 1,112 (0.008, 0.023) 30–45 2.3 565 (0.012, 0.039) 45–60 3.6 552 (0.022, 0.055) 60–75 8.4 407 (0.058, 0.115) 75–90 14.1 199 (0.096, 0.197) 90–100 28.6 14 (0.084, 0.581) Abbreviation:CI= confidence interval. Table 2. Predicted cross-species transmission risk levels and corresponding observed event probabilities for human–Suidae viruses.
Given its exceptionally high predicted zoonotic potential, we strongly recommend implementing enhanced surveillance protocols and strengthened biosecurity measures specifically targeting PCV3 to effectively mitigate potential spillover threats to human populations.
This study highlights that cross-species transmission of Suidae-associated viruses is jointly shaped by host genetic relatedness, viral genome features, and environmental factors. The BRT model demonstrated strong predictive performance and identified PCV3 as a virus with particularly high spillover risk. These findings provide a quantitative basis for early warning and surveillance prioritization and reinforce the importance of the One Health approach to prevent future zoonotic emergence.
-
Understanding and predicting cross-species transmission remains fundamental to pandemic preparedness. This study employed a BRT model to integrate viral, host, environmental, and anthropogenic variables for assessing spillover risk in human-Suidae zoonotic viruses. Our findings illuminate critical ecological and evolutionary drivers of zoonotic risk while establishing a data-driven framework for early warning and surveillance prioritization. Recent investigations have validated machine learning approaches for pathogen risk assessment (7-8), and our methodology advances this field by specifically targeting the human–Suidae transmission interface.
Host–human divergence time emerged as the most influential predictor in our analysis. This finding reinforces the principle that viruses demonstrate greater propensity for cross-species transmission between phylogenetically related species due to conserved receptor mechanisms and immunological similarities (5,9). Phylogenetic proximity facilitates viral entry, replication efficiency, and immune evasion strategies in novel hosts, thereby substantially increasing the likelihood of successful spillover events. Consequently, host evolutionary relationships should constitute a fundamental component of zoonotic risk assessment frameworks, particularly in biodiverse regions characterized by frequent human–animal interactions.
Viral genome size emerged as a strong predictor of spillover potential, consistent with previous studies demonstrating that larger genomes confer greater adaptive capacity and evolutionary flexibility (8). Environmental and anthropogenic factors — including rural human population density, precipitation patterns, and artificial surface coverage — proved significant predictors, reflecting both increased exposure opportunities and ecological disruptions that facilitate viral emergence. These findings underscore that zoonotic risk arises from complex interactions among viral characteristics, host biology, and human-mediated environmental changes (7).
Applying the BRT model to viruses not yet reported in humans highlighted Porcine circovirus 3 as a high-risk candidate, due to its stability, recombination capacity, and widespread distribution (10). Although no human infections have been reported, proactive surveillance of PCV3 and similar viruses is warranted, especially in regions with intensive Suidae farming and limited biosecurity. These results echo calls by Grange et al. (8) and Carlson et al. (7) for proactive identification of high-risk viruses before they emerge. We recommend that health authorities integrate such model outputs into zoonotic surveillance systems, prioritizing high-risk viruses like PCV3 for serological testing, metagenomic screening, and field investigations in both animal reservoirs and sentinel human populations. This predictive approach can help shift from reactive outbreak response to proactive early warning.
While these findings highlight the value of the BRT model for proactive surveillance, it is important to consider its limitations. First, the model depends on publicly available data, which may be geographically biased or incomplete. Sampling gaps, especially in low-resource or biodiverse regions, may affect model reliability (11). Second, spillover involves complex ecological and socio-behavioral processes not fully captured here. Nevertheless, the BRT framework is robust and could be integrated into zoonotic surveillance pipelines to guide evidence-based prioritization of high-risk viruses (12).
Our findings reinforce the fundamental One Health principle by demonstrating that viral characteristics, host biology, and environmental conditions collectively determine spillover risk rather than operating in isolation.
The integration of predictive modeling frameworks with comprehensive ecological and epidemiological surveillance systems can facilitate a paradigm shift from reactive outbreak response toward proactive risk management strategies, thereby enhancing global pandemic preparedness capabilities.
HTML
| Citation: |

|