
 Download:
Download:




-
Objective: Recent cholera outbreaks in Anhui Province have been linked to Vibrio cholerae O139, but information on these strains is limited. We established the first genomic dataset of local O139 strains to analyze the genomic characteristics and evolution of antibiotic resistance.
Methods: Thirty-four Vibrio cholerae O139 isolates from Anhui (2013–2024) were sequenced using next-generation sequencing. Genes for virulence, antimicrobial resistance, pathogenicity islands, and mobile genetic elements were predicted using ABRicate and other online tools. To construct a phylogenetic tree, 124 publicly available O139 genomes were included in the single-nucleotide polymorphism analysis alongside the study isolates.
Results: Strains formed two clusters that were genetically closer to China isolates than with those from Bangladesh and India. All strains harbored ctxA and ctxB, with partial deletions in virulence genes and pathogenicity islands; over 50% lacked vgrG-2 in the T6SS. Strains from 2022–2024 exhibited higher azithromycin but lower trimethoprim-sulfamethoxazole resistance than those collected during 2013–2017.
Conclusion: Vibrio cholerae O139 in Anhui are endemic to China, with limited virulence but strong colonization abilities. The increased azithromycin resistance rate may be driven by its clinical antimicrobial usage, suggesting its potential for continued antibiotic resistance evolution.
-
Vibrio cholerae serogroups O1 and O139 are known to cause cholera pandemics. Since 1817, seven global cholera pandemics have occurred, with the ongoing seventh pandemic beginning in 1961 (1). The global spread of O1 El Tor biotype has replaced the classical biotype as the cause of the current pandemic. In 1992–1993, the O139 serogroup first emerged, triggering outbreaks in India and Bangladesh, and was genetically derived from El Tor biotype strains (2-4). Unlike its widespread epidemics in Bangladesh and India, the O139 serogroup caused limited foodborne outbreaks in China (5). Nevertheless, recent studies indicate that O139 continues to cause sporadic outbreaks in this region. The first O139 cholera outbreak in China occurred in Xinjiang in 1993, with subsequent case reports from other provinces (6). Anhui Province detected its first O139 serogroup outbreak in 1996 (7). Although sporadic cases have persisted in the region, the genomic characteristics of these strains and their evolutionary relationships with isolates from other regions of China and Asia remain unclear. Antibiotics such as azithromycin (AZM), ciprofloxacin (CIP), and doxycycline are recommended for the clinical management of cholera (4). However, the recent emergence of antimicrobial resistance (AMR) in Vibrio cholerae has become an increasing concern. A study in China reported consistently high levels of resistance to streptomycin, trimethoprim-sulfamethoxazole (SXT), erythromycin, and polymyxin B among O139 serogroups from 1993 to 2009 (5). Since then, data on antibiotic resistance in these strains have been scarce.
Thus, to explore the genetic and antimicrobial features of Vibrio cholerae O139 in the Anhui Province, we analyzed 34 strains collected by the Anhui Provincial Center for Disease Control and Prevention from 2013 to 2024, sequenced their whole genomes, and assessed their drug resistance profiles.
Whole-genome sequencing was performed using MGISEQ2000 or Illumina NovaSeq platforms. Single-nucleotide polymorphisms (SNPs) were identified using Snippy (version 4.6.0; The University of Melbourne) and recombinant SNPs were removed using Gubbins (version 2.4.1; The Wellcome Trust Sanger Institute). The reference genome sequence was obtained from Bangladesh (GCF_900324445.1) (8). A maximum-likelihood phylogenetic tree was constructed using IQ-TREE (version 2.2.6; Australian National University). The AMR, virulence, and plasmid genes were predicted using ABRicate (version 1.0.1; The University of Melbourne) in the CARD, VFDB, and PlasmidFinder databases, respectively. Cholerae Finder v1.0 (https://cge.food.dtu.dk/services/CholeraeFinder/) was used to analyze the seventh pandemic islands (VSP-1 and VSP-2) and mobile genetic element (MGE) genes (intI 1 and intSXT). Antimicrobial susceptibility testing using broth microdilution for ampicillin (AMP), ampicillin sulbactam (AMS), ceftazidime (CAZ), cefotaxime (CTX), chloramphenicol (CHL), AZM, CIP, meropenem (MEM), amikacin (AMI), tetracycline (TET), and SXT was used to determine the minimum inhibitory concentration using a commercially prepared panel (Fosun Diagnostic, Shanghai, China). Results were interpreted according to the breakpoints defined by the Clinical and Laboratory Standards Institute. Escherichia coli ATCC25922 was used as the control.
Among the 34 strains, 33 were isolated from nine foodborne cholera outbreaks across six cities in Anhui Province, while one was identified through cholera surveillance in 2013. During the nine outbreaks, 28 cases were reported, including one in Jiangsu Province and another imported from Henan Province. Three outbreaks involved more than two cases; most individuals were asymptomatic. Epidemiological investigations revealed that six of the nine outbreaks were associated with banquets, and in four of these, turtles were the suspected food source. Twenty-seven strains were isolated from 26 cases. Three strains were isolated from turtle specimens, two of which were associated with outbreaks, and four strains were isolated from environmental samples, such as sewage, turtle cages, and closed stools, based on an epidemiological analysis (Table 1).
Strain City Isolation year Isolation source Clinical symptom Outbreak No. Suspected source No. of cases AH13HB006 Huaibei 2013 Turtle − − − − AH15TC021 Chuzhou 2015 Anal swab Asymptomatic carrier 1 Banquet/
Turtle2* AH15FY014 Fuyang 2015 Stool Confirmed case 2 Banquet/
Turtle1 AH15FY015 Fuyang 2015 Turtle − 2 AH15FY016 Fuyang 2015 Turtle cage − 2 AH15FY017 Fuyang 2015 Turtle cage − 2 AH17WXA018 Bozhou 2017 Stool Confirmed case 3 Unknown 1† AH22BZ152 Bozhou 2022 Stool Confirmed case 4 Banquet/
Turtle1§ AH22BZ153 Bozhou 2022 Stool Confirmed case 4 AH22BZ157-1 Bozhou 2022 Turtle − 4 AH22BZ154 Bozhou 2022 closestool − 4 AH22BB0202 Bengbu 2022 Anal swab Confirmed case 5 Banquet/
Unknown9¶ AH22BB0210 Bengbu 2022 Anal swab Asymptomatic carrier 5 AH22BB0209 Bengbu 2022 Anal swab Asymptomatic carrier 5 AH22BB0208 Bengbu 2022 Anal swab Asymptomatic carrier 5 AH22BB0207 Bengbu 2022 Anal swab Asymptomatic carrier 5 AH22BB0206 Bengbu 2022 Anal swab Asymptomatic carrier 5 AH22BB0201 Bengbu 2022 Anal swab Confirmed case 5 AH22BB0205 Bengbu 2022 Anal swab Asymptomatic carrier 5 AH23SZ0001 Suzhou 2023 Stool Confirmed case 6 Unknown 1 AH23SZ0002 Suzhou 2023 Sewage − 6 AH23BZ791 Bozhou 2023 Stool Confirmed case 7 Banquet/
Unknown1 AH23MAS0424 Maanshan 2023 Anal swab Asymptomatic carrier 8 Banquet/
Turtle11 AH23MAS0427 Maanshan 2023 Anal swab Asymptomatic carrier 8 AH23MAS0426 Maanshan 2023 Anal swab Asymptomatic carrier 8 AH23MAS0425 Maanshan 2023 Anal swab Asymptomatic carrier 8 AH23MAS0423 Maanshan 2023 Anal swab Asymptomatic carrier 8 AH23MAS0422 Maanshan 2023 Anal swab Asymptomatic carrier 8 AH23MAS0421 Maanshan 2023 Anal swab Confirmed case 8 AH23MAS0420 Maanshan 2023 Anal swab Asymptomatic carrier 8 AH23MAS0419 Maanshan 2023 Anal swab Asymptomatic carrier 8 AH23MAS0418 Maanshan 2023 Stool Asymptomatic carrier 8 AH23MAS0417 Maanshan 2023 Stool Confirmed case 8 AH24BB0141 Bengbu 2024 Stool Confirmed case 9 Shrimp 1 Note: *Two cases were associated with the same banquet in Anhui Province, and the other one was confirmed to have been found in Jiangsu Province.
† Imported case from Henan Province.
§ Two stool samples were collected from the same patient on different days.
¶ A total of 9 cases were associated with three separate banquets, and 8 of these cases were analyzed in this study.
“−” means not applicable.Table 1. Epidemic information of the 34 Vibrio cholerae O139 isolates.
A total of 1,118 SNPs were identified across our isolates and 124 publicly available Vibrio cholerae O139 sequences from India, Bangladesh, and China. Of these, 870 SNPs were located on chromosome I and 248 on chromosome II. The phylogenetic tree divided the isolates into three lineages. All isolates from this study, along with 90 from Zhejiang, and 9 from Shanghai, were classified under lineage 3. Thirty-one isolates from this study were distributed across two clusters within lineage 3. Cluster 1 consisted of four isolates from a 2015 outbreak and was supported by eight SNPs. Cluster 2 consisted of 27 isolates from six outbreaks from 2022–2023, supported by 11 SNPs. One environmental isolate from the 2023 outbreak showed significant evolutionary divergence from its associated case isolate, differing by 181 SNPs. Three additional isolates were found outside of the two clusters (Figure 1).
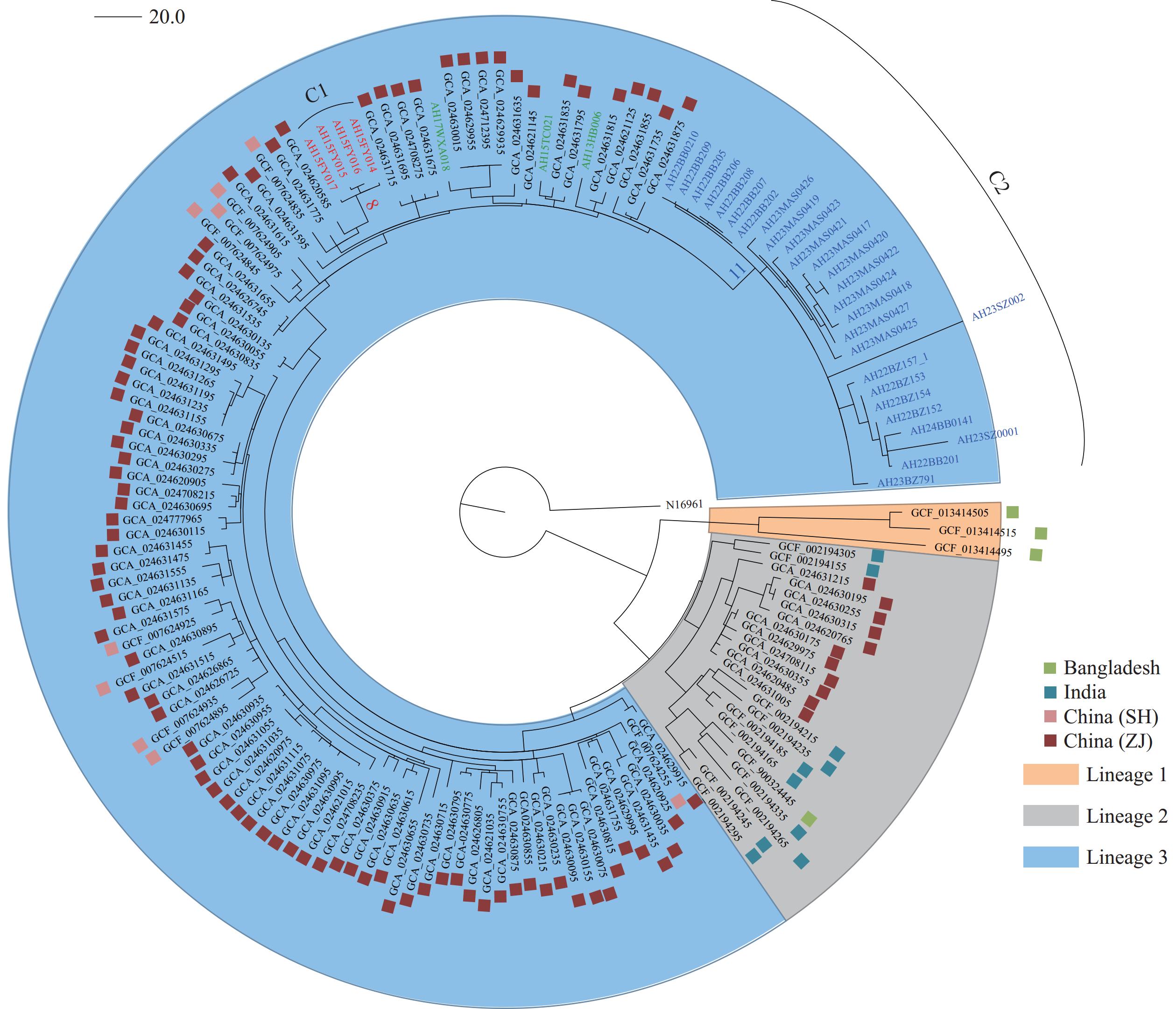 Figure 1.
Figure 1.Maximum-likelihood phylogenetic tree of 158 Vibrio cholerae O139 isolates. The tree was rooted using the seventh pandemic O1 strain N16961 as the outgroup.
Note: The clusters C1 and C2 are depicted in red and blue, respectively. The number of SNPs supporting each cluster is marked on the respective branches. Three strains not belonging to either cluster are shown in green.
Abbreviation: SH=Shanghai; ZJ=Zhejiang.
All 34 isolates harbored ctxA and ctxB, whereas five lacked additional CTX phage genes. The majority also harbored additional toxin genes and various pilus-encoding genes, such as pilA and MSHA genes. Variable deletions were observed within the T6SS and pathogenicity islands, with vgrG-2 deleted in >50% of the isolates (Figure 2A). Thirteen AMR genes were highly prevalent (>88%) among the study isolates, whereas sul1, dfrA18, and qacE delta1 were detected only in isolates collected before 2017. The genes mphE, mphF, and msrE were primarily found in Cluster 2, whereas aph (3’)-Ia, aadA2, dfrA12, and tetD were mainly associated with Cluster 1 (Figure 2B). All isolates contained intSXT, with 32 isolates carrying the IncA/C plasmid; however, only seven isolates collected from 2013–2017 contained intI 1 (Figure 2A).
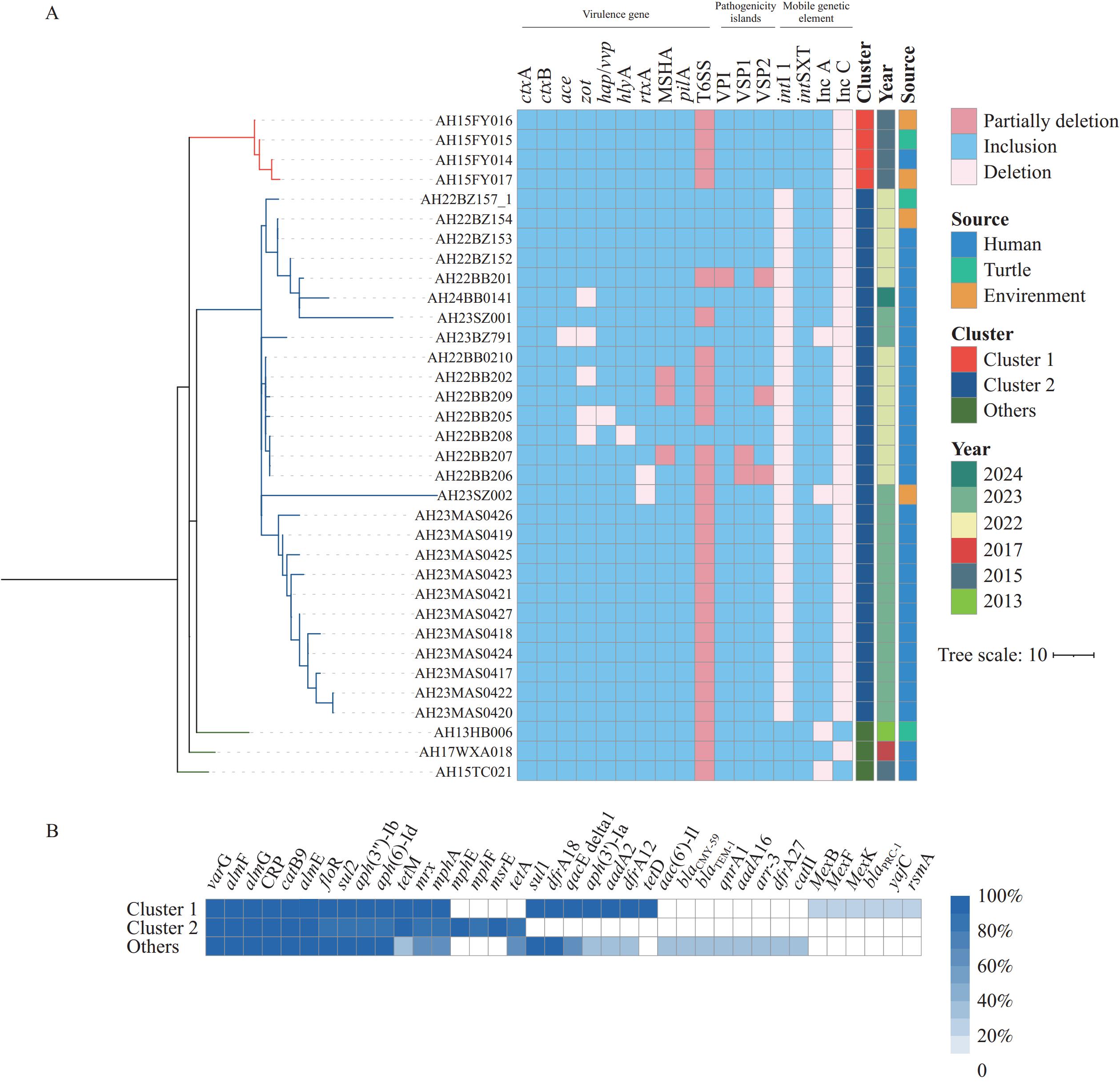 Figure 2.
Figure 2.Genomic features of the 34 Vibrio cholerae O139 isolates. (A) Virulence genes, pathogenicity islands, and mobile genetic elements. (B) Distribution of antibiotic resistance genes across the different clusters.
Note: For (A), MSHA gene cluster includes mshHIJKLMNEGF and mshBACD. Both T6SS and VPI comprise 19 genes, as previously described (11). For (B), ‘Others’ represents three isolates that fall outside the two main clusters.
Abbreviation: MSHA=maltose-sensitive haemagglutinin.
All isolates were susceptible to MEM, CTX, CAZ, and AMI, with the majority being susceptible to AMP and AMS. Notably, high resistance rates to TET (91.18%) and AZM (76.55%) were observed, with AZM resistance particularly high among isolates collected during 2022–2024. All isolates collected during 2013–2017 exhibited 100% SXT resistance, whereas this was significantly lower (3.7%) among isolates collected between 2022 and 2024. Drug resistance patterns demonstrated a clear temporal trend, with TET-AZM resistance predominant in strains collected between 2022–2024 and TET-SXT resistance in those collected during 2013–2017 (Table 2).
Antibiotic 2013–2017 (n=7) 2022–2024 (n=27) S (%) I (%) R (%) S (%) I (%) R (%) CHL 4 (57.1) 2 (28.6) 1 (14.3) 10 (37.0) 17 (63.0) 0 SXT 0 0 7 (100) 26 (96.3) 0 1 (3.7) MEM 7 (100) 0 0 27 (100) 0 0 CTX 7 (100) 0 0 27 (100) 0 0 CTZ 7 (100) 0 0 27 (100) 0 0 TET 2 (28.6) 1 (14.3) 4 (57.1) 0 0 27 (100) CIP 6 (85.7) 1 (14.3) 0 27 (100) 0 0 AZM 7 (100) 0 0 1 (3.7) 0 26 (96.3) AMI 7 (100) 0 0 27 (100) 0 0 AMP 6 (85.7) 0 1 (14.3) 27 (100) 0 0 AMS 6 (85.7) 0 1 (14.3) 27 (100) 0 0 Abbreviation: AMP=ampicillin; AMS=ampicillin sulbactam; CAZ=ceftazidime; CTX=cefotaxime; CHL=chloramphenicol; AZM=azithromycin; CIP=ciprofloxacin; MEM=meropenem; AMI=amikacin; TET=tetracycline; SXT=trimethoprim-sulfamethoxazole; S=susceptible; I=intermediate; R=resistant. Table 2. Antibiotic susceptibility patterns of the 34 Vibrio cholerae O139 isolates.
-
In recent decades, foodborne outbreaks caused by Vibrio cholerae O139 with soft-shelled turtles as vectors have been frequently reported in China (9). This study established the first provincial-level genomic surveillance dataset for this pathogen in Anhui, addressing the critical data gap. From 2013 to 2024, all cholera cases in Anhui Province were attributed to the serogroup O139. Group dining and human mobility were identified as the key outbreak drivers, with phylogenetic analysis implicating turtles as transmission vehicles. The isolates showed closer genetic clustering with the Zhejiang and Shanghai strains than with other Asian strains, suggesting domestic circulation rather than cross-border transmission. Two temporally segregated clusters were identified within the Anhui isolates, demonstrating the ongoing evolution of Vibrio cholerae O139 in China. This pattern differs from three genetically distinct but temporally overlapping O139 clades that emerged in India and spread across South Asia between 1992 and 2015 (10).
Most isolates harbored multiple virulence genes but exhibited more deletions than Zhejiang O139 isolates (11) and those from other Asian countries (10), potentially affecting virulence. T6SS plays a crucial role in host-pathogen interactions via translocation. Deletion of vgrG-2 or both vgrG1 and vgrG3 severely impairs T6SS secretion in Vibrio cholerae (12). Thus, the absence of vgrG-2 in the study isolates might impair T6SS function, potentially explaining the limited cases and asymptomatic infections in Anhui Province. However, high prevalence of MSHA genes may enhance their environmental viability, particularly on the surface of turtles, where MSHA is essential for colonization (9).
In China, the resistance rates to TET, CHL, AZM, and AMP in toxigenic O139 strains from 1993 to 2009 varied over time. Notably, TET resistance increased significantly in 1998 and remained high, whereas CHL, AZM, and AMP resistance decreased after an initial increase (5). Substantial temporal variations in AMR were observed among isolates. AZM resistance increased, whereas SXT susceptibility was retained, possibly owing to shifts in antibiotic treatment patterns. In the 1970s, SXT was the primary treatment for cholera, but AZM and CIP have become the preferred therapy in recent clinical practice (4,13). Antimicrobial pressure may have contributed to the acquisition of macrolide-resistance genes, including mphE and mphF. A gradual loss of AMR genes with the wave progression of O139 in other Asian countries may explain the decline of this lineage (10). However, our analysis revealed that local O139 strains acquired new AMR genes, particularly those associated with AZM resistance, which enabled continuous transmission in the region.
The study has two primary limitations. First, the analysis was restricted to O139 isolates from a single province from 2013 to 2024, with limited samples. Second, the limited recent global O139 genomic data could have reduced phylogenetic resolution, and these findings may not fully represent the genetic diversity or evolutionary dynamics of O139 strains across broader regions. Despite low pathogenicity, the Anhui O139 strains demonstrated robust colonization and resistance gene transmission, indicating persistent local transmission and the emergence of new resistance patterns.
HTML
| Citation: |

|