
 Download:
Download:




-
The first acquired immune deficiency syndrome (AIDS) case in China was reported in 1985 (1), marking the beginning of HIV-1 spread throughout the country. According to the National Statutory Infectious Disease Epidemic Profile published by the National Bureau of Disease Control and Prevention (NBDCP) in 2023, China documented 59,533 AIDS cases and 22,393 related deaths (https://www.ndcpa.gov.cn/). Although the implementation of immediate identification and treatment strategies has significantly reduced HIV incidence, HIV-1 remains a substantial threat to public health in China.
In Anhui Province, the identification of the first human immunodeficiency virus-positive (HIV-positive) case in 1994 was followed by a rapid increase in infections, particularly among commercial blood donors (2). Anhui has served as a crucial hub in the dissemination of HIV-1 across China (3). By 2018, the province had documented 17,183 HIV-infected individuals. The predominant HIV-1 subtypes circulating in Anhui include CRF01_AE, CRF07_BC, CRF08_BC, CRF55_01B, and subtype B. Previous studies have revealed a significant prevalence of unique recombinant forms (URFs), indicating frequent superinfections. The ongoing recombination between subtypes enhances HIV genetic diversity and facilitates the emergence of highly adaptive variants that could potentially trigger new pandemics (4). Given the substantial subtype diversity in Anhui, continuous monitoring of subtype distributions remains essential for effective HIV containment (5). Furthermore, surveillance of HIV genetic characteristics through near-full length genome (NFLG) analysis provides crucial insights into the HIV epidemic in Anhui and informs the development of innovative strategies for preventing and treating new HIV infections.
-
The sampling strategy in Anhui Province employed a risk-stratified approach, categorizing prefectural-level cities into high, medium, and low risk based on reported HIV cases over the preceding three years. The sampling framework mandated the inclusion of at least two cities from each risk category, with minimum sampling proportions of 20%, 40%, and 60% for high-, medium-, and low-risk cities, respectively.
The study encompassed 238 newly confirmed HIV-1 positive samples collected from Anhui in 2023. Plasma samples were procured through the Anhui Provincial CDC.
-
Viral RNA was extracted from plasma samples using the QIAamp Viral RNA Mini Kit (QIAGEN, Germany). HIV-1 cDNA synthesis and amplification were performed using the AccessQuick™ RT-PCR System (PROMEGA, China) in combination with 2× Taq PCR Mix (TIANGEN, China). The amplification and sequencing of HIV genome fragments were conducted according to previously established protocols (6-7).
Sequencing was performed on an ABI 3730XL sequencer (Applied Biosystems, USA). Sequence processing, including trimming, splicing, and mixed base interpretation, was conducted using Sequencher v5.4.6 (Gene Codes Corporation, Mississippi, USA). To ensure data quality and exclude experimental cross-contamination, sequences were validated using the WHO HIVDR QC Tool. Multiple sequence alignment was performed using MAFFT v7 (Research Institute for Microbial Diseases Osaka University, Osaka, Japan) (8). Reference sequences were obtained from relevant databases, and phylogenetic analysis was conducted using IQ-TREE (version 2.0, University of Vienna, Vienna, The Republic of Austria; The Australian National University, Canberra, Australia) (9) with the Maximum Likelihood (ML) method, implementing the General Time Reversible (GTR) + G nucleotide substitution model and 1,000 bootstrap replicates. The resulting phylogenetic trees were visualized using the interactive tree of life (iTOL) online platform (https://itol.embl.de/).
-
Near full-length genome sequences were obtained using a two-amplicon strategy following previously established protocols. The methodologies for viral RNA extraction, PCR amplification, and sequencing procedures have been detailed in previous publications (10).
Sequence analysis was performed using Sequencher software (version 5.4.6, Gene Codes Corporation, USA) for sequence trimming, secondary peak detection, sequence assembly, and mixed base interpretation. Quality control of the assembled sequences was conducted using the online Gene Cutter tool (https://www.hiv.lanl.gov/content/sequence/GENE_CUTTER/cutter.html). To eliminate potential contamination, all sequences were compared against the comprehensive HIV sequence database using the Basic Local Alignment Search Tool (BLAST) (http://hiv-web.lanl.gov/content/index). Sequences meeting quality control criteria were analyzed for potential unique recombinant patterns using two complementary approaches: the Recombinant Identification Program (RIP) with default parameters (https://www.hiv.lanl.gov/content/sequence/RIP/RIP.html) and the jumping profile Hidden Markov Model (jpHMM) (http://jphmm.gobics.de/) within the HIV Database.
Based on the RIP and jpHMM analyses, we constructed a comprehensive reference sequence alignment incorporating predominant subtypes circulating in China (CRF01_AE, CRF07_BC, CRF08_BC, CRF55_01B, and subtype B), other Chinese CRFs, and relevant external sequences. Sequence alignment was performed using Aliview software (Uppsala University, Upsala, Sweden) (11). Phylogenetic analysis was conducted using IQ-TREE (version 2.0, University of Vienna, Vienna, The Republic of Austria; The Australian National University, Canberra, Australia) (9) with the GTR model and 1,000 bootstrap replicates. The resulting phylogenetic trees were visualized using the iTOL online platform. Recombination breakpoint analysis was performed using SimPlot (version 3.5.1, Biomatters, Auckland, New Zealand) (12) with parameters set to a 300 bp window size and 20 bp step size. The genetic structure of unique recombinant samples was mapped using the Recombinant Mapping Tool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html) available in the HIV Database.
-
From the total cohort of 238 newly reported cases in Anhui Province, 37 cases (15.5%) demonstrated subtype inconsistencies across at least two of the three analyzed genomic regions: PR-RT (nucleotides 2253-3500, HXB2), IN (nucleotides 4230-5096, HXB2), and env (nucleotides 7016-7650, HXB2). Among these 37 samples exhibiting subtype discordance, seven contained sufficient plasma volume for NFLG amplification and sequencing, ultimately yielding 5 complete NFLG sequences. These 5 sequences were confirmed as URFs and designated as AHAQ230009, AHBZ230009, AHBZ230031, AHCUZ230011, and AHXC230017. The demographic characteristics of these 5 individuals are detailed in
Supplementary Table S1 . -
Comprehensive phylogenetic analysis was performed on three distinct genomic regions: PR-RT, IN, and env. Reference sequences were obtained from HIV databases, and maximum likelihood (ML) phylogenetic trees were constructed using IQ-TREE (version 2.0), implementing the GTR + G nucleotide substitution model with 1,000 bootstrap replicates. Tree visualization was accomplished using the iTOL online web tool. The subtype classifications for the three regions (PR-RT, IN, and env) corresponding to the five sequences are summarized in Figure 1 and Table 1.
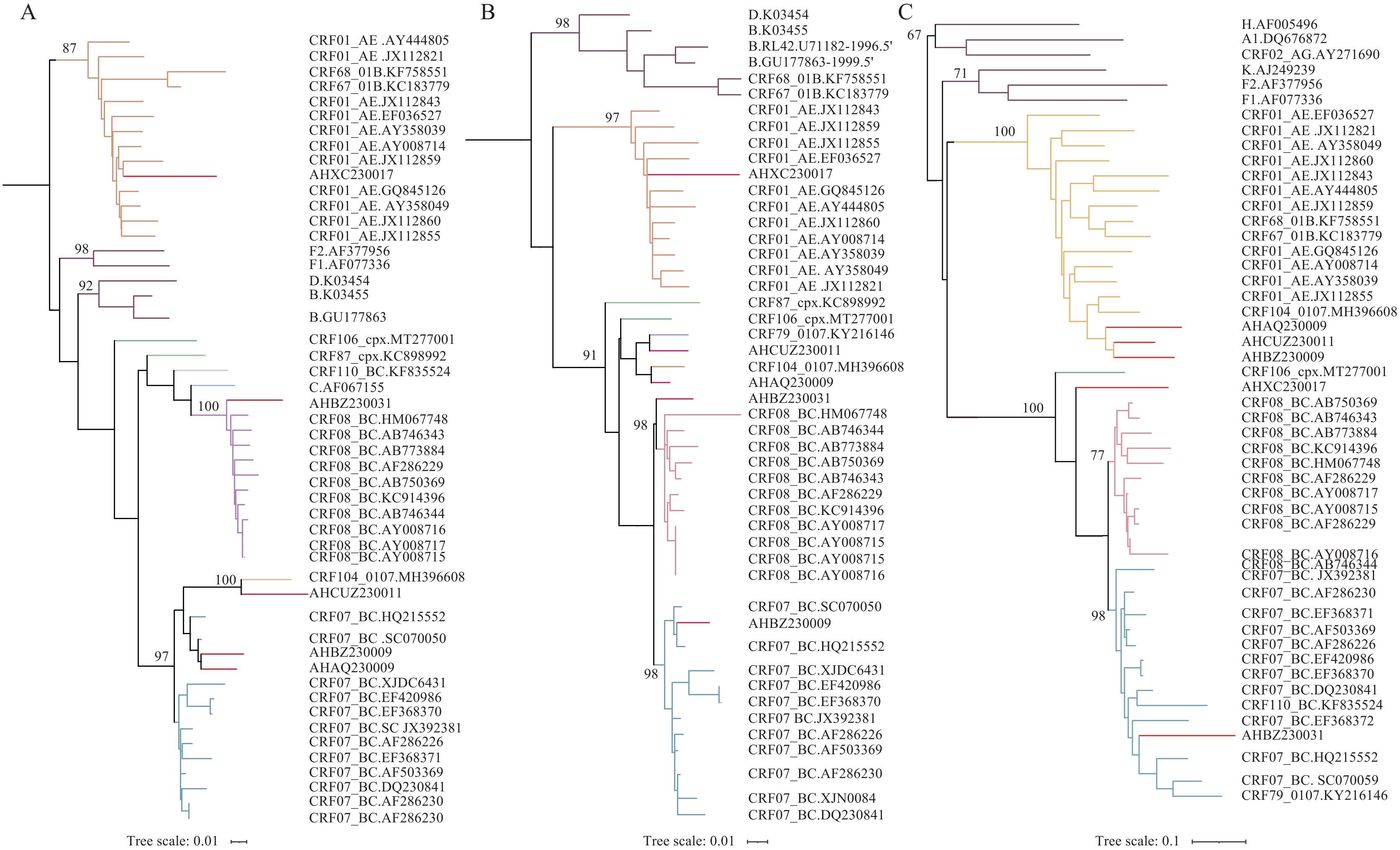 Figure 1.
Figure 1.Phylogenetic tree of three fragments of five samples. (A) PR-RT; (B) IN; (C) env.
Note: Phylogenetic tree produced by IQ-TREE, the reliability of the tree branches was assessed by 1,000 bootstrap replicates, and trees were visualized by iTOL. The red line represents the five samples (AHAQ230009, AHBZ230009, AHBZ230031, AHCUZ230011 and AHXC230017).
Abbreviation: PR-RT=protease-reverse transcriptase; IN=integrase; env=envelope; iTOL=theinteractive tree of life.
Sample ID Subtype of PR-RT Subtype of IN Subtype of env AHAQ230009 CRF07_BC CRF104_0107 CRF01_AE AHBZ230009 CRF07_BC CRF07_BC CRF01_AE AHBZ230031 CRF08_BC CRF08_BC CRF07_BC AHCUZ230011 CRF104_0107 CRF79_0107 CRF01_AE AHXC230017 CRF01_AE CRF01_AE URF Abbreviation: PR-RT=protease-reverse transcriptase; IN=integrase; env=envelope; URF=unique recombinant forms. Table 1. Subtypes of three fragments (PR-RT, IN, env) of five samples.
-
The sequences obtained from the five potential URFs exceeded 8,000 base pairs in length, encompassing the complete coding regions for gag, pol, vif, vpr, vpu, rev, tat, env, and nef genes. Phylogenetic analysis conducted using IQ-TREE2 (Figure 2) demonstrates that these five samples represent distinct lineages independent of previously identified CRFs.
 Figure 2.
Figure 2.Phylogenetic tree of five NFLGs.
Note: The sequences of the potential URFs are marked in red. The neighbor joining tree of five NFLGs was constructed using IQ-TREE 2, and the reliability of the tree branches was assessed by 1,000 bootstrap replicates, and trees were visualized by iTOL.
Abbreviation: NFLGs=near-full length genome; URF=unique recombinant forms; iTOL=the interactive tree of life.
Detailed analysis revealed specific breakpoint locations within the HXB2 genome reference framework for all five sequences. As shown in Figure 3, the mosaic structures of these recombinants are characterized as follows: AHAQ230009 comprises three segments: ⅠCRF07_BC (634-4884); ⅡCRF01_AE (4885-8404); and ⅢCRF07_BC (8405-8960). AHBZ230009 consists of three regions: ⅠCRF07_BC (634-6323); ⅡCRF01_AE (6324-8327); and ⅢCRF07_BC (8328-9552). AHBZ230031 exhibits a more complex structure with seven segments: ⅠCRF07_BC (620-1933); ⅡCRF08_BC (1934-3001); ⅢCRF07_BC (3002-3215); ⅣCRF08_BC (3216-4628); ⅤCRF07_BC (4629-8518); ⅥCRF08_BC (8519-9098); and ⅦCRF07_BC (9099-9470). AHCUZ230011 displays six distinct regions: ⅠCRF07_BC (608-2959); ⅡCRF01_AE (2960-4244); ⅢCRF07_BC (4245-4885); ⅣCRF01_AE (4886-8327); ⅤCRF07_BC (8328-8811); and ⅥCRF01_AE (8812-9354). AHXC230017 shows six segments: ⅠCRF01_AE (621-4832); ⅡB (4833-4899); ⅢCRF01_AE (4900-6937); ⅣCRF07_BC (6938-7679); ⅤB (7680-8240); and ⅥCRF01_AE (8241-9552) (
Supplementary Table S2 ). The phylogenetic analyses for all identified fragments are presented inSupplementary Figure S1 .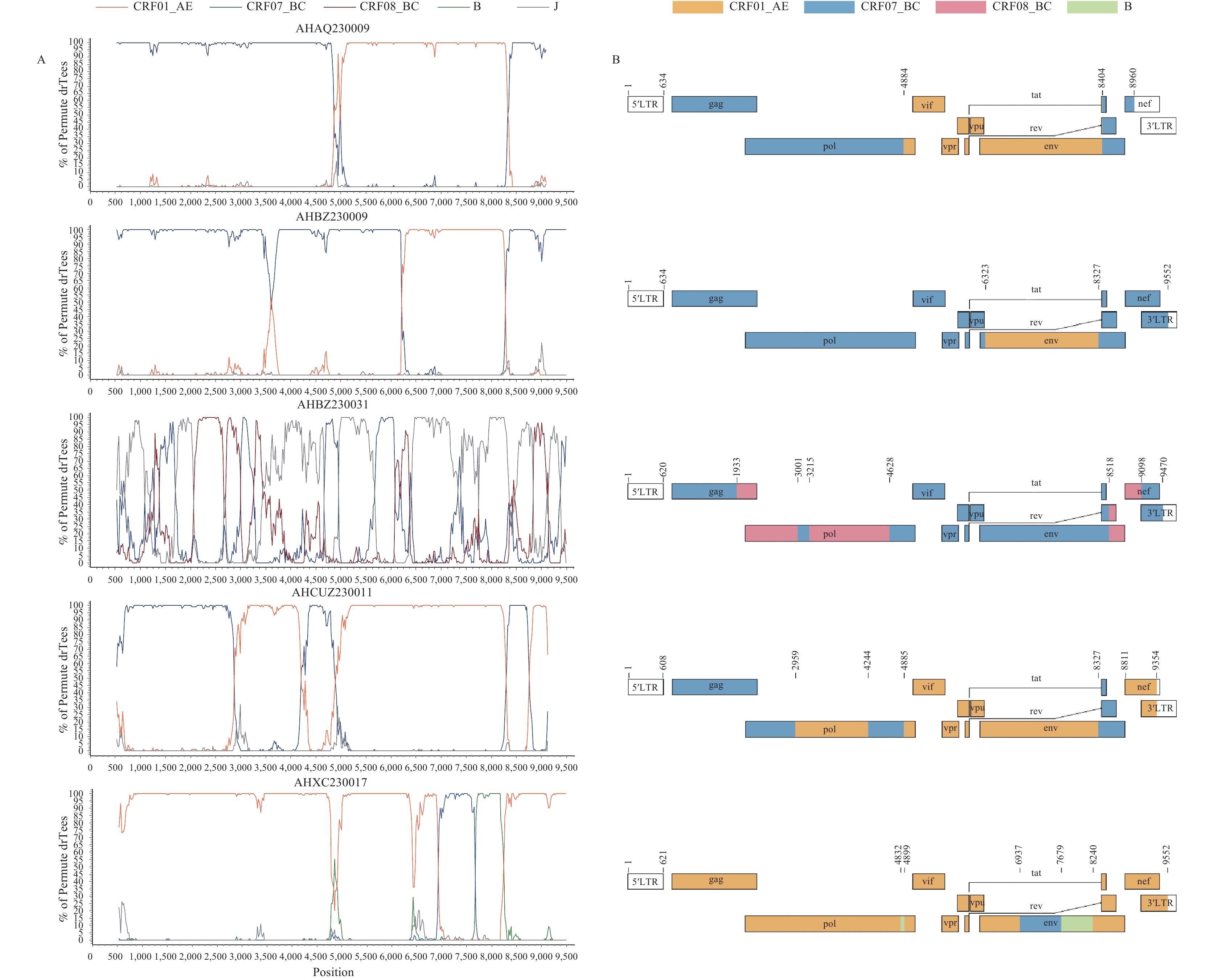 Figure 3.
Figure 3.Recombinant analysis of the five identified URFs. (A) Bootscan analyses of the five URFs; (B) Genome map of the five URFs.
Note: (A) Bootscan analyses of the five URFs were performed using a window size of 300 bases and a step size of 20 bases, GapStrip: on, Kimura (2-parameter), T/t: 4.0. The CRF01_AE reference group includes JX112859, EF036527,and AY358049. The subtype CRF07_BC reference group includes SC070050, HQ215552 , AF286230. The subtype CRF08_BC reference group includes KC914396 and AY008715. The subtype B reference group includes GU177863 and U71182. The subtype J reference is SE9280. The x-axis represents the nucleotide positions, while the y-axis of the bootscan analysis shows the percentage bootstrap values of the permuted trees. (B) The analysis of recombination breakpoints in the five URFs. The mosaic fragments in the near-full-length genomes (NFLGs) recombinants are color-coded as follows: CRF01_AE (orange), CRF07_BC (blue), CRF08_BC (pink), and subtype B (green). The nucleotide positions of each fragment are numbered according to the HIV-1 reference sequence HXB2 (K03455).
Abbreviation: URF=unique recombinant forms.
Bootscan analysis (Figure 3A) and similarity plot (
Supplementary Figure S2 ) provide substantial evidence that these five isolates represent unique recombinants, distinct from previously identified CRFs or URFs. The recombinant structures of the five near-full-length sequences were delineated based on the breakpoint locations (Figure 3B), thereby confirming their classification as novel URFs. -
Analysis of drug resistance mutations revealed that none of the five NFLGs exhibited resistance-associated mutations in either the protease or integrase regions. However, the AHXC230017 sequence contains the V179D mutation in the reverse transcriptase region, which is associated with non-nucleoside reverse transcriptase inhibitors. This mutation conferred intermediate-level resistance to Efavirenz (EFV) and Nevirapine (NVP), while demonstrating low-level resistance to Rilpivirine (RPV).
-
In this study, we identified five URFs in Anhui Province through comprehensive NFLG analysis. These URFs exhibited distinct recombination patterns, characterized by mosaic segments derived from multiple viral subtypes distributed throughout their genomes. While current CRF classification primarily relies on NFLG sequence analysis (13-14), this approach offers significant advantages over shorter gene fragment analysis (such as PR-RT, IN, and env), particularly in delineating recombination patterns and precise breakpoint locations. This methodology provides crucial insights for HIV-1 molecular epidemiological surveillance.
Recent years, the rapid dissemination of HIV-1 CRF07_BC strain among sexually transmitted populations in China leads to the emergence of numerous variants, including both URFs and novel CRFs (15). Our finding that CRF07_BC participated in all five URF recombination events suggests its enhanced transmission capability and infection prevalence (1).
Anhui Province, strategically located in central China and bordered by Jiangsu, Zhejiang, and Shanghai, serves as a major labor-exporting inland province and has emerged as a significant hub for HIV transmission and spread (3). The co-circulation of multiple subtypes in this region increases the probability of inter-subtype recombination (16), potentially challenging existing prevention and treatment strategies. Therefore, vigilant monitoring of novel URFs is essential for effectively evaluating and providing early warning of emergent strains with enhanced transmission potential.
However, the relatively small sample size of this study limits its contribution to monitoring the epidemic situation across the entire province. Furthermore, this study employed Sanger sequencing, which may not identify occurrences of superinfection or drug resistance mutations in certain samples. Future efforts should concentrate on increasing the sample size and utilizing more advanced sequencing techniques such as Nanopore technology to analyze the samples, thereby obtaining more comprehensive information.
In conclusion, our identification of five URFs through near full-length HIV-1 genome analysis not only expands our understanding of HIV-1 genetic diversity but also underscores the critical importance of NFLG sequencing in molecular surveillance efforts.
-
The staff of the Anhui Provincial CDC for their assistance with sample collection and processing.
-
All participants provided informed consent. Approved by the Ethics Committee of the National Center for AIDS/STD Control and Prevention, China CDC (approval number X140617334).
HTML
Study Population
Sequencing and Analysis of PR-RT, IN, and env Fragments
Sequencing and Analysis of Near Full-length Genome
Demographic Information of the Study Population
Phylogenetic Analysis of Genomic Regions
Near Full-length Genome Analysis
Drug Resistance Analysis
| Citation: |

|