
 Download:
Download:




-
Gastrointestinal infections, predominantly foodborne, are among the most common illnesses worldwide, affecting an estimated 550 million people annually, including 220 million children younger than five years of age (1). Salmonella enterica is a major pathogen implicated in these infections. Salmonella comprises more than 2,500 serovars. Salmonella Typhimurium and Salmonella Enteritidis are frequently associated with diarrheal illnesses (2). Recent reports indicate that outbreaks can also be attributed to rare Salmonella serovars. For example, S. Ealing, S. Welikade, S. Isangi, and S. Coeln have been linked to outbreaks in the UK, France, China, and Norway, respectively (3–6).
One Health is an integrated approach to balancing and optimizing the health of people, animals, and ecosystems. It leverages the close, interdependent links among these fields to create new surveillance and disease control methods (7). Healthy individuals, particularly food handlers, could be potential sources of hidden Salmonella dissemination. Therefore, monitoring Salmonella prevalence among healthy individuals is essential for comprehensively understanding and managing the associated risks.
Monitoring the prevalence of multidrug-resistant (MDR) Salmonella isolates is a common method for assessing the risk of salmonellosis. This approach facilitates timely detection and intervention in cases of salmonellosis, thereby limiting further pathogen dissemination. Whole-genome sequencing (WGS) is widely used to predict and identify serovars, virulence factors, and antimicrobial resistance genes (ARGs) in Salmonella surveillance.
Using resistance profiling and genomic analysis of Salmonella isolates collected from healthy individuals during a decade-long, large-scale, laboratory-based investigation, we identified five rare Salmonella serovars reported for the first time in China and revealed the prevalence of MDR isolates and the hidden dissemination of these serovars.
Between 2013 and 2020, fecal samples were systematically collected from individuals undergoing occupational health examinations in Yulin City, Guangxi Zhuang Autonomous Region, China. Salmonella spp. were cultured from these samples according to a previously established protocol (8). Serovars were determined using slide agglutination and by predicting genome sequences using Salmonella In Silico Typing Resource (SISTR). A systematic literature review using keyword searches for “Salmonella” and “serotype” was performed. Corresponding genomes were retrieved from the EnteroBase and GenBank databases to verify the novelty of the identified serovars within China.
Two commercial antimicrobial susceptibility testing panels (BD Phoenix™ NMIC-413 and BD Phoenix™ RUONMIC-801) were used with the microbroth dilution method, following the manufacturer’s instructions. For clarity, only the common panel of 11 antibiotics is presented, namely: Amikacin (AMK), Ceftazidime (CAZ), Chloramphenicol (CHL), Ciprofloxacin (CIP), Colistin (COL), Ertapenem (ETP), Meropenem (MEM), Piperacillin-Tazobacta (TZP), Tetracycline (TET), Tigecycline (TIG), Trimethoprim-Sulfamethoxazole (SXT). Results were interpreted in accordance with the Clinical and Laboratory Standards Institute (CLSI) 2022 guidelines. Escherichia coli ATCC 25922 served as the quality control strain.
Genomic DNA was extracted from 15 bacterial isolates using the Wizard Genomic DNA Purification Kit (Promega) after incubation. Paired-end libraries with insert sizes of 300-500 bp were constructed and sequenced on the MGI200 platform according to the manufacturer’s protocol. The sequencing reads for each isolate were assembled using SPAdes v3.5.0. ARGs, plasmid profiles, and virulence factors were identified using Abricate software and databases, including CARD, PlasmidFinder, and VFDB.
To trace the origins of the S. Ouakam and S. Ealing serotype epidemics, 79 S. Ouakam and 114 S. Ealing isolates were sourced from the EnteroBase database. These isolates were analyzed using single-nucleotide polymorphisms (SNPs) and hierarchical clustering of core genome multilocus sequence typing (HierCC). The analyses were performed using the online analytical pipeline within EnteroBase. The resulting data were used to generate a phylogenetic tree and visualized using the iTOL v6 platform.
A total of 7,044 Salmonella isolates were recovered from 372,708 individual samples in a retrospective investigation conducted from 2013 to 2022, with an isolation rate of 1.89% among healthy individuals. Among these, 119 distinct Salmonella enterica serovars were identified in healthy individuals from Yulin. Utilizing an extensive literature review, we report the first identification of five rare Salmonella serovars in China. A total of 14 isolates of rare serovars were sourced from individuals ranging in age from 17 to 60 years, comprising 86% (12/14) males and 14% (2/14) females. Notably, 93% (13/14) of the individuals were engaged in food handling, while the remaining 7% (1/14) were not. The isolates comprised five rare serovars: S. Welikade (n=1), S. Mountpleasant (n=1), S. Coeln (n=1), S. Ealing (n=2), and S. Ouakam (n=9).
S. Welikade, S. Mountpleasant, and S. Ealing isolates were susceptible to all 11 antibiotics tested. In contrast, the S. Coeln isolate and nine S. Ouakam isolates were MDR. Both exhibited resistance to CHL, SXT, and TET. Notably, the S. Coeln isolate was resistant to TIG, an antibiotic typically reserved for severe Gram-negative infections. Meanwhile, all nine S. Ouakam isolates were resistant to CAZ and CIP (Table 1).
Strain Serovars Year of isolation Antimicrobial resistance profile Sa13427 Salmonella Welikade 2013 − Sa15171 Salmonella Mountpleasant 2016 − Sa17519 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17528 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17531 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17537 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17538 Salmonela Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17539 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17541 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17542 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa17544 Salmonella Ouakam 2020 CHL-SXT-TET-CAZ-CIP Sa20877 Salmonella Coeln 2022 CHL-SXT-TET-TIG Sa21032 Salmonella Ealing 2022 − Sa21220 Salmonella Ealing 2022 − Note: “−” indicates isolates with no antimicrobial resistance profile. Table 1. Antimicrobial resistance profile of rare serovar isolates from Yulin City, Guangxi Zhuang Autonomous Region, China.
Analysis of the 15 isolates’ genomes revealed five distinct plasmid types. The two S. Ealing isolates lacked plasmids, whereas the S. Mountpleasant isolate harbored an IncQ1 plasmid. The S. Welikade isolate contained both Col440I and p0111 plasmids. All S. Ouakam and S. Coeln isolates carried the IncHI2/IncHI2A plasmid (Figure 1).
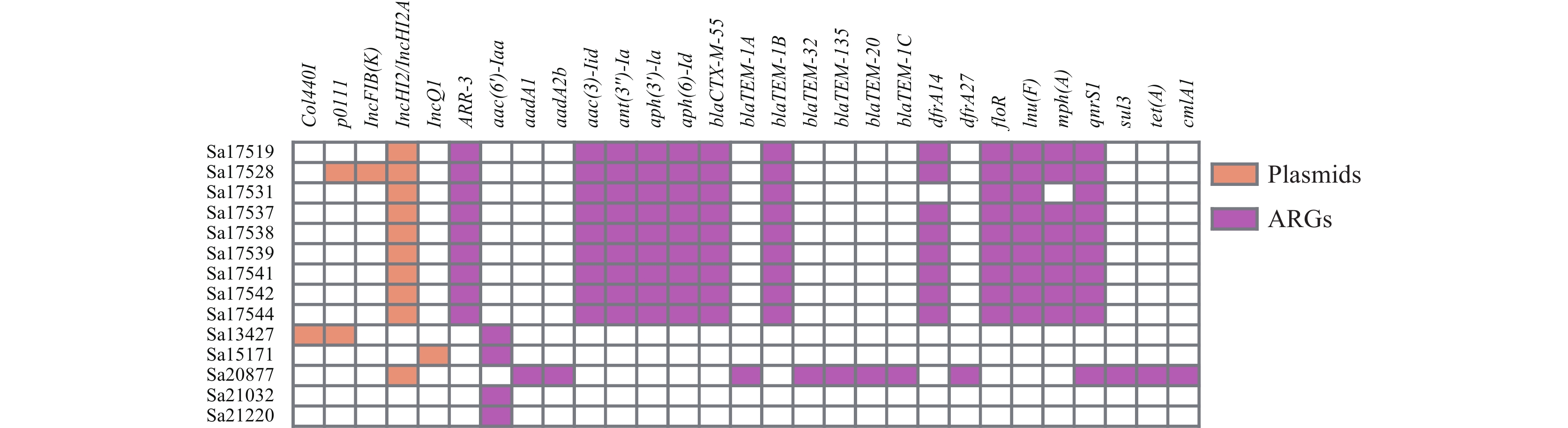 Figure 1.
Figure 1.Distribution of plasmids and antimicrobial resistance genes (ARGs) among the studied isolates.
The S. Ouakam isolates possessed diverse ARGs, conferring resistance to aminoglycosides, sulfonamides, lincosamides, tetracyclines, beta-lactams, chloramphenicol, quinolones, and rifamycins. The S. Coeln isolates exhibited resistance mediated by a broader spectrum of 14 ARGs, attributed to the presence of IncHI2/IncHI2A plasmids. Without these plasmids, the S. Welikade, S. Mountpleasant, and S. Ealing isolates displayed a more restricted resistance profile, predominantly featuring a single ARG, aac(6')-Iaa (Figure 1).
Our investigation revealed 147 distinct virulence genes within the Salmonella isolates, highlighting multiple mechanisms underlying virulence and pathogenicity. The cdtB gene for typhoid toxin production was found in S. Welikade, while astA (EAST1 toxin) was detected in the Sa17631 isolate of S. Ouakam. Furthermore, all rare serotype isolates harbored gene clusters involved in iron uptake (entA, entB, entC, entD, entE, entF, entS) and the mig-14 gene, associated with resistance to antimicrobial peptides (Figure 1).
We analyzed a dataset of 34,408 SNPs to elucidate the phylogenetic relationships among 88 S. Ouakam genomes. The resulting phylogenetic tree revealed two distinct groups (Figure 2A). To facilitate further analysis, we reconstructed the phylogenetic tree specifically for the isolates of group 2 (Figure 2B). Nine S. Ouakam isolates, derived from healthy individuals, formed a cluster within clade α, demonstrating SNP distances ranging from 0 to 58. To define outbreak clusters accurately, we employed the internationally recognized HierCC HC5 standard, which is widely used in infectious disease outbreaks (9). Seven S. Ouakam isolates classified under the same HC5 cluster (type 428586) were obtained from individuals employed by the same company, indicating the potential for hidden transmission of Salmonella spp. among this population.
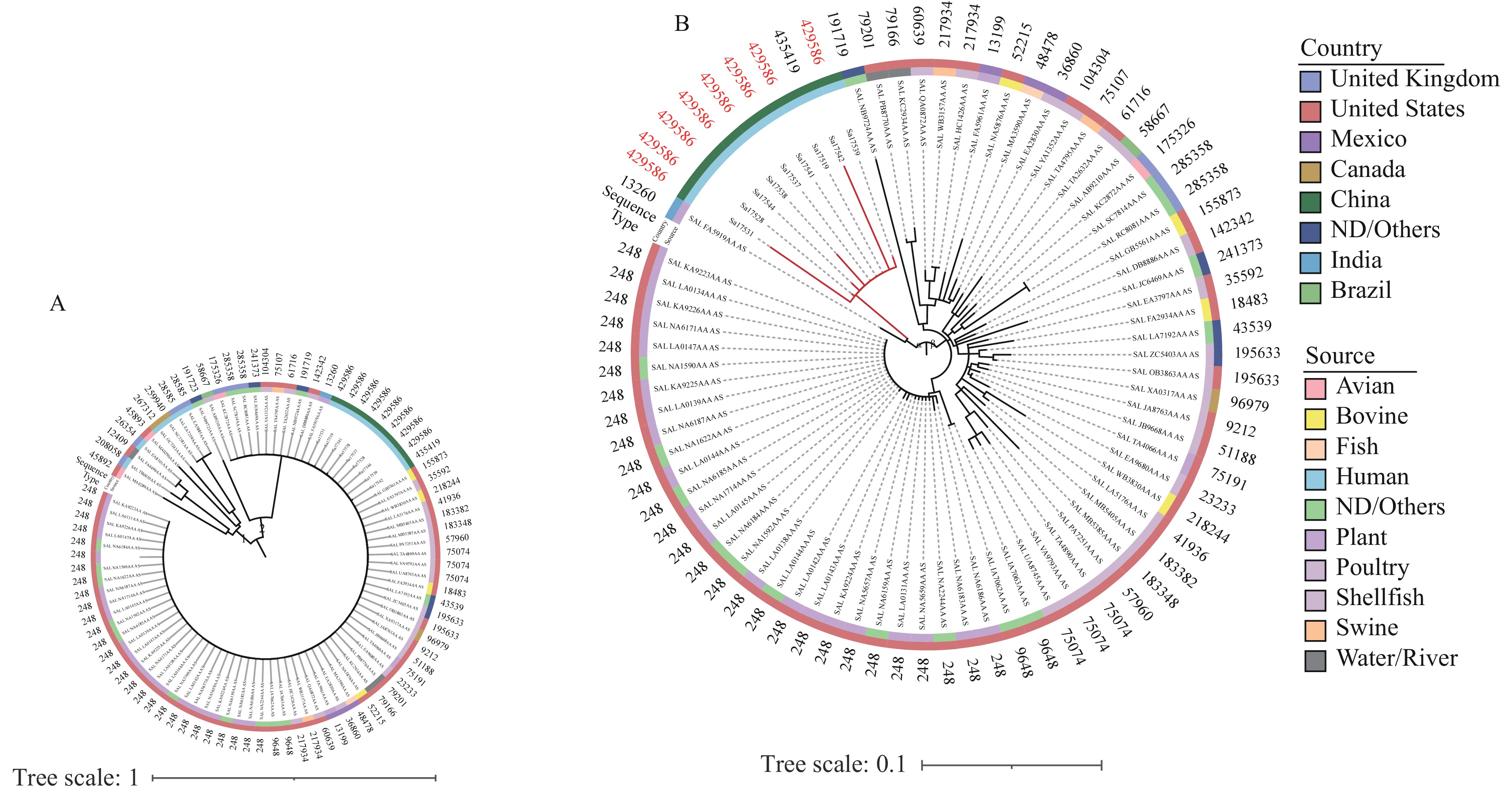 Figure 2.
Figure 2.Phylogenetic tree of Salmonella Ouakam isolates. (A) Phylogenetic structure of 88 Salmonella Ouakam isolates, including 79 isolates from EnteroBase and 9 isolates from Yulin. (B) Phylogenetic tree of isolates belonging to group 2 in Figure 2A.
Note: The isolates in red font represent those collected in this study.Analysis of 116 isolates allowed for phylogenetic tree reconstruction for S. Ealing, revealing two distinct groups. Within group B, two clades, designated α and β, were identified. Isolates recovered in this study, characterized by HC5 type 435357, clustered within clade α. Two additional isolates within clade α, originating from plants in Paraguay and the Netherlands, corresponded to HC5 types 268261 and 214748, respectively. The remaining four isolates within clade α were all characterized by HC5 type 353819 (Figure 3).
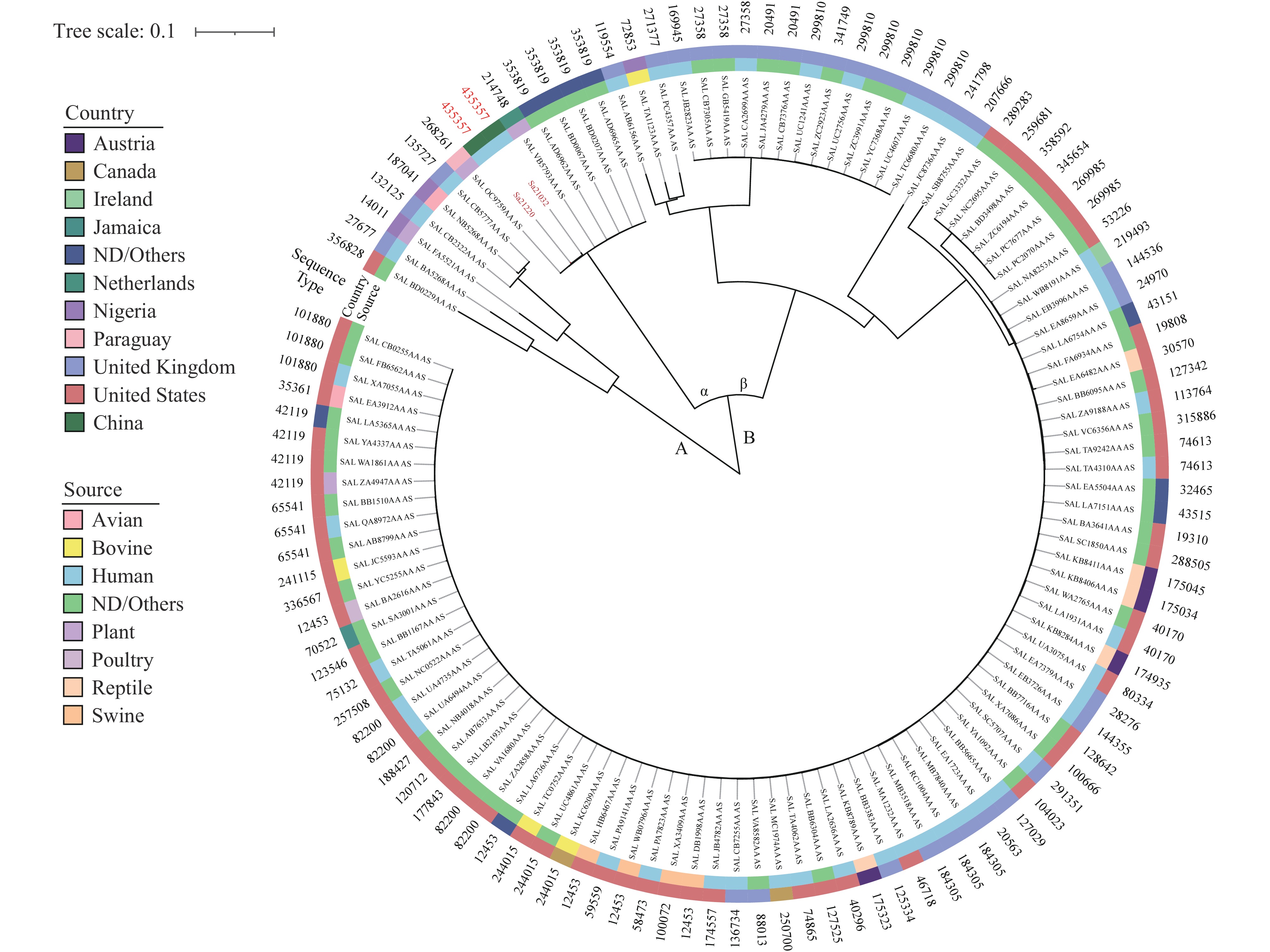 Figure 3.
Figure 3.The phylogenetic tree of 116 S. Ealing isolates, including 114 isolates from EnteroBase and 2 isolates from Yulin City, Guangxi Zhuang Autonomous Region, China.
Note: The isolates collected from Yulin are highlighted in red. -
From 2014 to 2021, the top 10 serovars among sporadic clinical Salmonella isolates in China were Typhimurium, Enteritidis, I 4,[5],12:i:-, London, Stanley, Derby, Rissen, Thompson, Agona, and Goldcoast. In southern China, Typhimurium and I 4,[5],12:i:- were the most prevalent serovars (10). Although none of the five rare serovars identified in this study have been found in clinical isolates in China, previous reports have documented the presence and dissemination of rare Salmonella serovars across various geographic locations, impacting both agricultural and public health sectors. S. Ealing has been identified in turkey production in the USA (11), within food samples in Nigeria (12), and among poultry in Brazil (13). In 1985, an outbreak of S. Ealing affecting infants was reported in the UK (3). Outbreaks of S. Coeln linked to minced meat and salad consumption were recorded in France in 1998 and Norway from 2013 to 2014, respectively (6). Data on S. Welikade are scarce, with isolated reports from France, Sri Lanka, Australia, and Sweden (4). No reports concerning S. Mountpleasant have been identified. Furthermore, S. Ouakam isolates have been sourced from a variety of contexts, including ground turkey, turkey breakfast sausage (14), retail poultry, pigs, the swine environment (15), and chicken meat (16) from children under five with diarrhea in rural Burkina Faso (17). This study is the first to elucidate the genomic characteristics linked to virulence and resistance in five rare Salmonella serovars isolated from healthy individuals in China. Through analysis of these genomic features, we aim to contribute evidence to improve risk assessment and deepen understanding of the determinants of antimicrobial resistance and pathogenicity among these emerging pathogens.
Seven S. Ouakam isolates were collected from healthy individuals, all employees at the same enterprise, within one week. Furthermore, classification within the same HC5 cluster suggests potential hidden, asymptomatic Salmonella transmission. The isolation of S. Ealing strains of the same HC5 type nearly one month apart suggests that this serovar may exhibit relative stability over a short period, indicating the potential for sustained transmission and the presence of asymptomatic carriers, which increases the risk of spread. Therefore, monitoring transmission dynamics and detecting asymptomatic carriers are crucial for controlling its spread and preventing disease.
This study was subject to at least two limitations. First, the food strains, which asymptomatic carriers may come into contact with, were not collected, so the transmission of strains from food to humans could not be determined. Second, there were no detailed epidemiological data to confirm an outbreak of S. Ouakam among the asymptomatic employees. Over a 10-year surveillance period, we documented the first domestic identification of five rare Salmonella serovars. Genomic sequencing and phenotypic resistance profiling revealed that these rare serovar isolates were MDR and harbored ARGs. Phylogenetic analysis further revealed the undetected dissemination of S. Ouakam. This study underscores the critical importance of active surveillance among healthy individuals, especially within key occupational groups such as food workers, to monitor foodborne pathogens.
HTML
| Citation: |

|