
 Download:
Download:




-
Monkeypox virus (MPXV), a member of the Orthopoxvirus genus within the Poxviridae family, is a double-stranded DNA virus measuring approximately 200–250 nm with a genome of approximately 197 kb (1). It is categorized into two main branches: Branch I (Central Africa) and Branch II (West Africa). Branch I viruses demonstrate higher pathogenicity, transmission capability, and mortality rates than Branch II viruses (2). Furthermore, Branch II is subdivided into sub-branches IIa and IIb, with IIb being the predominant genotype in the global monkeypox outbreaks since 2022 (3).
In late May 2023, the first locally acquired case of monkeypox in Beijing, linked to imported cases, spread to other provincial-level administrative divisions (PLADs) in China. By 31 December 2023, there were 1,712 confirmed cases across 29 PLADs (4). To elucidate the genetic characteristics of MPXVs in Anhui Province since June 2023, we sequenced and analyzed the whole genomes of seven samples to identify the specific genetic alterations and variation patterns of MPXV in this region.
The samples were collected from laboratory-confirmed monkeypox patients and sent to the Anhui Provincial Center for Disease Control and Prevention by municipal Centers for Disease Control and Prevention in 2023, in accordance with the “Monkeypox Prevention and Control Technical Guidelines (2022 Edition).” These strains underwent whole-genome sequencing using the MGISEQ-2000 from MGI Tech Co. and the MinION platform from Oxford Nanopore Technologies. Subsequent analysis of nucleotide changes, amino acid mutations, and phylogeny was performed using MAFFT (version 7), IQ-TREE (version 2), and the online genotyping platform Nextclade (https://clades.nextstrain.org/).
Compared with the reference sequence (NC_063383.1), there were 91 single nucleotide polymorphisms (SNPs) distributed across the inverted terminal repeats (ITRs), variable region, and central region of MPXV. This included 49 (53.85%) GA>AA mutations and 34 (37.36%) TC>TT mutations (Figure 1A and 1B). These GA>AA and TC>TT nucleotide substitutions are attributed to hypermutations mediated by Apolipoprotein B mRNA Editing Catalytic Polypeptide-like 3 (APOBEC3), which facilitate the rapid transmission of MPXV (5). Of the 91 SNPs, 42 were non-synonymous mutations, 36 were synonymous mutations, and 13 were intergenic mutations. The 42 non-synonymous mutations were primarily found in the OPG093, OPG145, OPG176, and OPG210 genes, with 8 affecting viral proteins, 13 impacting host regulation, and 21 related to virus replication or transcription (Figure 1C and 1D). Comparison revealed that 7 sequences shared 77 mutations, 8 mutations were partially shared, and 6 mutations were unique. The SNPs of 2023AH-005 and 2023AH-006 were identical, and those of 2023AH-002, 2023AH-003, and 2023AH-007 differed by fewer than 5 from the preceding 2 sequences. There were 2 SNP differences between 2023AH-001 and 2023AH-004 (Figure 1E).
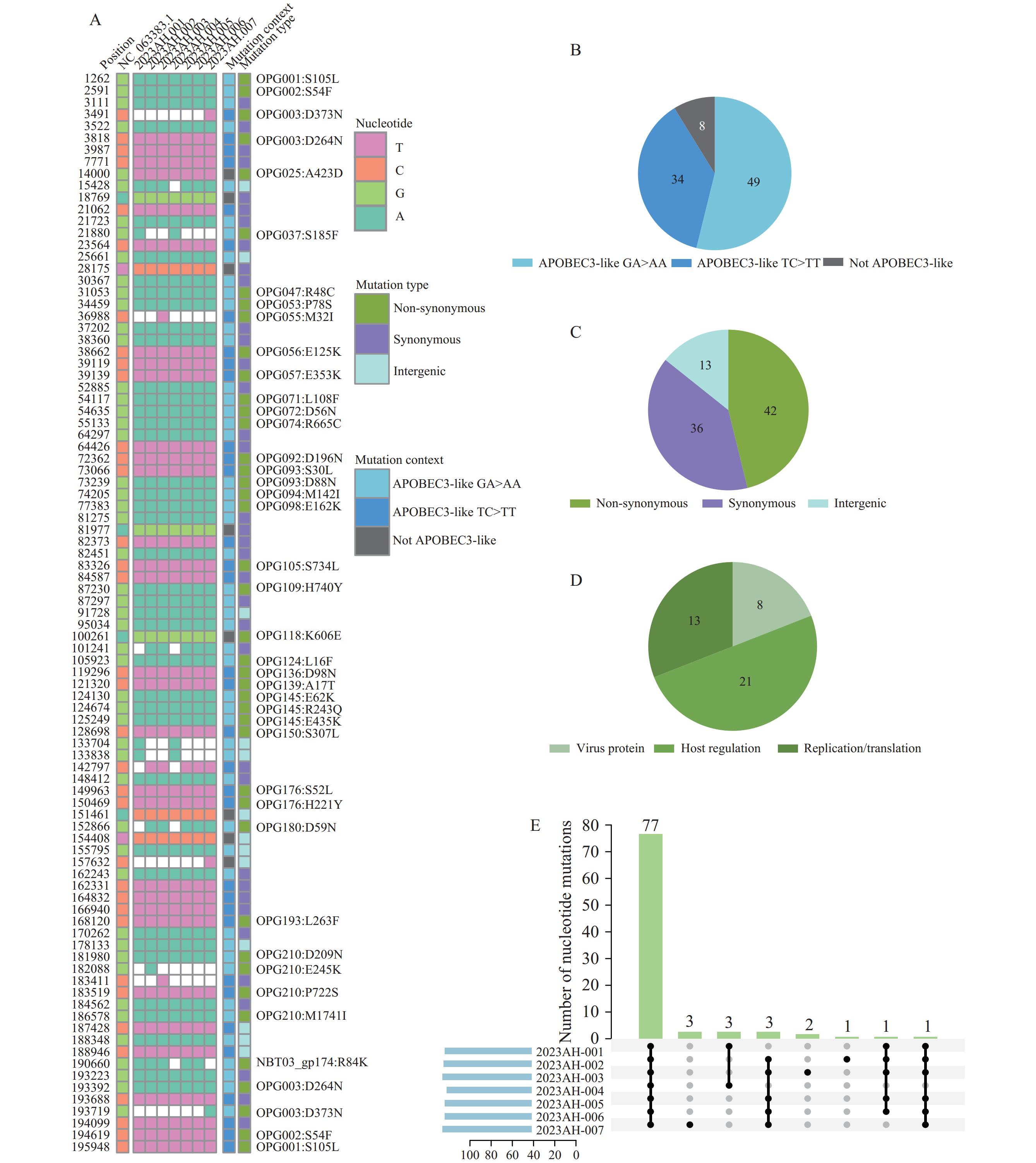 Figure 1.
Figure 1.Molecular evolution characteristics of seven local MPXVs from Anhui 2023. (A) Compared to the reference gene (NC_063383.1); (B) Distribution of nucleotide substitutions for the 91 SNPs; (C) Types of mutations among the 91 SNPs; (D) Distribution of 42 non-synonymous mutations; (E) Comparison of mutations between sequences.
Note: Panel A shows the Anhui monkeypox sequence contained 91 SNPs.
Abbreviation: SNP=single nucleotide polymorphisms; MPXV=monkeypox virus.
As of June 30, 2024, a total of 1,967 whole MPXV genome sequences were retrieved from the GISAID monkeypox database based on the criteria of “complete,” “high coverage,” “low coverage exclusion,” and “complete date of collection .” Five MPXVs from each branch and lineage were randomly selected for inclusion in the dataset; if fewer than 5 were available, all were included. A total of 396 sequences, including all high-quality, complete MPXV of lineages IIb C.1 and IIb C.1.1 from the GISAID and NCBI databases (with duplicate sequences removed) and the 7 sequences in this study, were integrated into the dataset. These MPXV genomes were aligned using MAFFT v7.511 software, and the alignment was trimmed using trimAL v1.4 software. Subsequently, a maximum likelihood phylogenetic tree was constructed using the best-fit K3Pu+F+I substitution model with IQTREE v2.3.6, and node support was calculated by the ultrafast bootstrap approximation method with 1,000 repetitions. The analysis revealed that the sequences from this study belonged to the same lineage IIb C.1.1 as the MPXV strains isolated in Guangdong, Yunnan, Jiangsu, and Zhejiang of China, as well as Japan, America, and Portugal. This cluster was distinct from the strains introduced into China earlier (Chongqing: EPI_ISL_15005641; Hong Kong Special Administrative Region: EPI_ISL_14945299; and Taiwan, China: EPI_ISL_13632071) (6–8). Furthermore, the sequences in this study could be divided into two clusters. 2023AH-001 and 2023AH-004 showed high similarity with sequences from Zhejiang, Yunnan, and Jiangsu PLADs of China, and the Republic of Korea and Japan, including the TMIPH0076 (EPI_ISL_17692269) sequence reported in Japan, which was related to the imported cases reported in Beijing (9). The remaining 5 sequences demonstrated high similarity with sequences from Guangdong of China, America, Portugal, and the Netherlands (Figure 2).
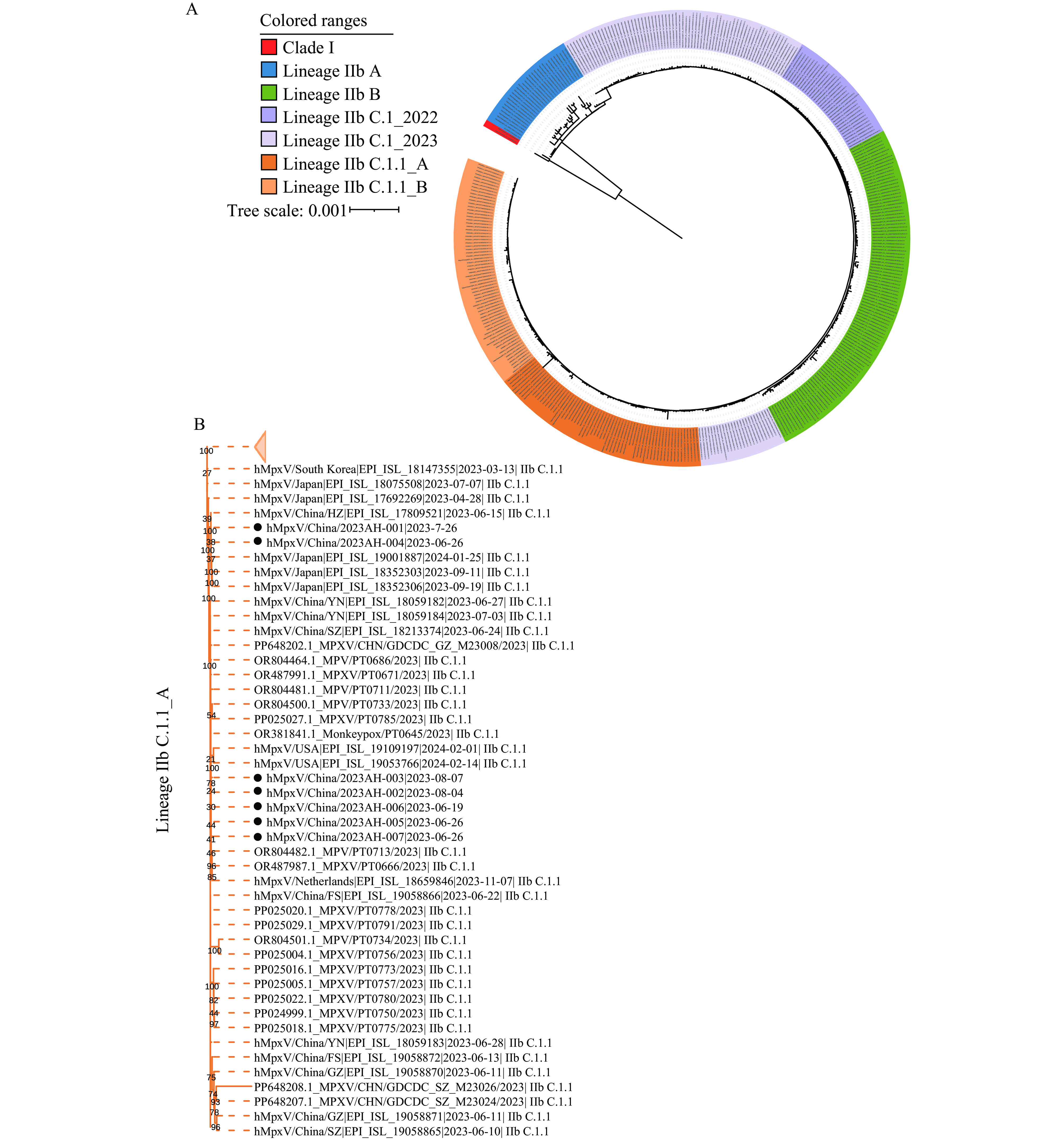 Figure 2.
Figure 2.Phylogenetic analysis of MPXV genome sequences associated with the local Anhui outbreak in 2023. (A) Phylogenetic analyses were based on 396 whole genome sequences of MPXV; (B) The phylogenetic characteristics of lineage IIb C.1.1_A including all 7 MPXV from Anhui.
Note: In panel A, The tree labels are colored by each lineage; the tree is rooted using clade I as an outgroup. In panel B, the black circle represents gene sequences in this study. -
Compared with the reference sequence (NC_063383.1), this study identified 91 SNPs in the MPXV sequences. Among these, 49 and 34 were characterized as APOBEC3-like GA>AA and TC>TT variations, respectively, representing 91.21% of the total mutations. This is consistent with the established evolutionary mechanism mediated by APOBEC3 protein in MPXV, where adaptive point mutations serve as the evolutionary mode, resulting in the formation of numerous microevolutionary branches (5). The MPXV sequences analyzed featured 36 synonymous and 42 non-synonymous mutations. The synonymous mutations, although not altering amino acids, contribute to codon deoptimization of MPXV, potentially associated with a decrease in mortality rates (10). The non-synonymous mutations, affecting viral proteins, host immune regulation, and viral replication transcription regions, impacted host immune responses and viral function. For instance, shared mutations in OPG002, OPG003, and OPG25 regions interfere with interferon functions such as TNF-α and IL-1, aiding in viral immune escape. Private mutations include OPG003:D373N involved in immune regulation, OPG037:S185F involved in apoptosis, and OPG055:M32I involved in cell movement (11). These private mutations, specific to individual cases, likely emerged following multiple transmissions, indicating covert dissemination of MPXV within Anhui Province and microevolution in genes related to immune evasion and pathogenicity.
Phylogenetic analysis indicated that the MPXV genomes in this study belonged to the IIb C.1.1 lineage and formed two clusters. The 2023AH-001 and 2023AH-004 sequences closely resembled those of strains from Zhejiang Province in China, Japan, and the Republic of Korea, while the remaining five sequences closely matched those from Guangdong, America, Portugal, and the Netherlands. These results, consistent with SNP differences among the seven sequences, suggest that the monkeypox cases in Anhui Province may represent multiple transmission chains. Furthermore, they differ from earlier reported MPXV in China and share close genetic similarities with the strains from Japan and the Repubic of Korea; the TMIPH0076 strain from Japan is linked to the first local case in Beijing (9). Conversely, based on genomic cluster analysis, MPXVs belonging to the C.1.1 lineage from PLADs such as Anhui, Guangdong, Yunnan, and Zhejiang are potentially linked to Portugal. Portugal, among the first countries to report cases in the 2022 MPXV outbreak, has experienced multiple viral lineage evolution events (12). However, without further epidemiological evidence, determining whether these seven MPXV sequences originated overseas or from interprovincial transmission within China remains challenging.
This study has several limitations. First, the poor cooperation of monkeypox patients, who often concealed their true situation, resulted in a lack of accurate epidemiological evidence. Only 7 cases of monkeypox in Anhui Province were sequenced and analyzed during the early stages, so the transmission chain of MPXV in Anhui Province could not be accurately determined. Second, there are few whole genome sequences of MPXV in China in public databases, making it impossible to determine the complete transmission network and comprehensively analyze the microevolution characteristics of MPXV in China.
In conclusion, this study demonstrates that the MPXVs identified in Anhui differed from the initially reported imported MPXV strains in China and early-stage MPXVs in China but belonged to the same IIb C.1.1 lineage as those appearing concurrently in other parts of China, with multiple transmission chains and no new branches. However, microevolutionary changes in areas related to viral replication, transcription, and immune escape pose risks of new local secondary outbreaks and covert transmissions. Meanwhile, the World Health Organization declared on 14 August 2024 that a more lethal strain of MPXV was rapidly spreading in Africa, with the potential to spread further to other continents. This announcement again categorized the monkeypox epidemic as a public health emergency of international concern. Thus, ongoing genomic surveillance of MPXVs is essential to fully understand the evolutionary trajectory and transmission patterns of MPXV in humans and to develop effective prevention strategies.
HTML
| Citation: |

|