
 Download:
Download:




-
Campylobacter jejuni (C. jejuni) is widely recognized as the primary causative agent of global foodborne bacterial diarrheal disease (1). Despite a limited number of documented C. jejuni outbreaks in China (2), a young patient in Yunnan Province presented with suspected tularemia. The patient presented with high fever and convulsions rather than typical diarrheal symptoms, but the isolated strain was biochemically identified as C. jejuni.
To elucidate the genetic features contributing to the clinical phenotype in this case, the isolate underwent comprehensive whole-genome sequencing. This investigation aimed to discern the genetic relationships between this strain and previously sequenced C. jejuni strains, considering population structure, differences in genomic composition, and common virulence factors in comparison to reference strains. Additionally, the presence of specific virulence factors was investigated. Furthermore, 13 major components of the Type VI Secretion System (T6SS) and Campylobacter jejuni Integrated Element 3 (CJIE3) were identified and analyzed.
A 20-month-old boy was admitted to the hospital with a maximum temperature of 40 °C, which had fluctuated around 39 °C for eight hours prior. The patient exhibited no respiratory or gastrointestinal symptoms but experienced a single, one-minute convulsion five minutes prior to admission, after which he regained consciousness. The initial diagnosis was herpes pharyngitis with febrile convulsions. The patient had no history of infection. His white blood cell count was within the normal range at 10.8×109/L (with elevated neutrophils at 78.1% and decreased lymphocytes at 14.0%). Hypersensitive C-reactive protein levels were elevated at 41.5 mg/L. Procalcitoninogen and erythrocyte sedimentation rate were both within normal limits at 0.134 ng/mL and 11 mm/h, respectively. Electroencephalogram and digital computed tomography scan of the head and lungs revealed no abnormalities. Unexpectedly, blood culture detected Francisella tularensis using the VITEK 2 Compact System based on the positive PyrA. Intracranial infection and central nervous system disease were ruled out based on the patient’s signs and symptoms and clinical test results. The study was approved by the Ethics Committee of the Yunnan Institute of Endemic Diseases Control & Prevention (Ethics Committee approval number 2021-09). Informed consent was obtained from legal guardians.
In the reported clinical case of suspected tularemia, blood was inoculated onto cysteine heart agar blood plates suitable for F. tularensis in a 5% CO2 incubator at 37 °C. After 48 hours of incubation, the culture medium exhibited several single colonies, 1–2 mm in diameter, with a teardrop shape and consistent morphology. Tularemia was ruled out in this patient based on negative tests for F. tularensis-specific antigens and antibodies. The colonies were Gram-stain-negative and elongated in an S-shape without spores, and all were identified as C. jejuni through positive reactions for oxidase, catalase, and hippurate hydrolysis. One strain was selected for high-quality whole-genome sequencing and confirmed as C. jejuni. This strain, designated L8, was tested for susceptibility to 12 antibiotics. The results showed that L8 was resistant to ciprofloxacin and tetracycline and sensitive to the other antibiotics. Comparisons using ResFinder (
https://cge.food.dtu.dk/services/ResFinder/ ) and CARD (https://card.mcmaster.ca/analyze ), a database of drug resistance genes, revealed that L8 expressed cmeABC and cmeR, which are closely related to macrolide, fluoroquinolone, cephalosporin, and fusidane resistance. The tetracycline, doxycycline, and minocycline resistance gene tet(O) was also expressed.The C. jejuni L8 genome was completely sequenced (GenBank accession no. CP139640) and comprised a single contig of 1,732,398 bp with no plasmids (Figure 1A). The L8 genome exhibited a G+C content of 30.29% and an ANI of 98.44%. Although L8 shared similar genomic characteristics with NCTC 11168, collinear analyses indicated that L8 possessed an additional fragment of 93,816 bp (Figures 1C, 1D), housing approximately 90 more genes, of which 55 had functional annotations (
Supplementary Table S1 ).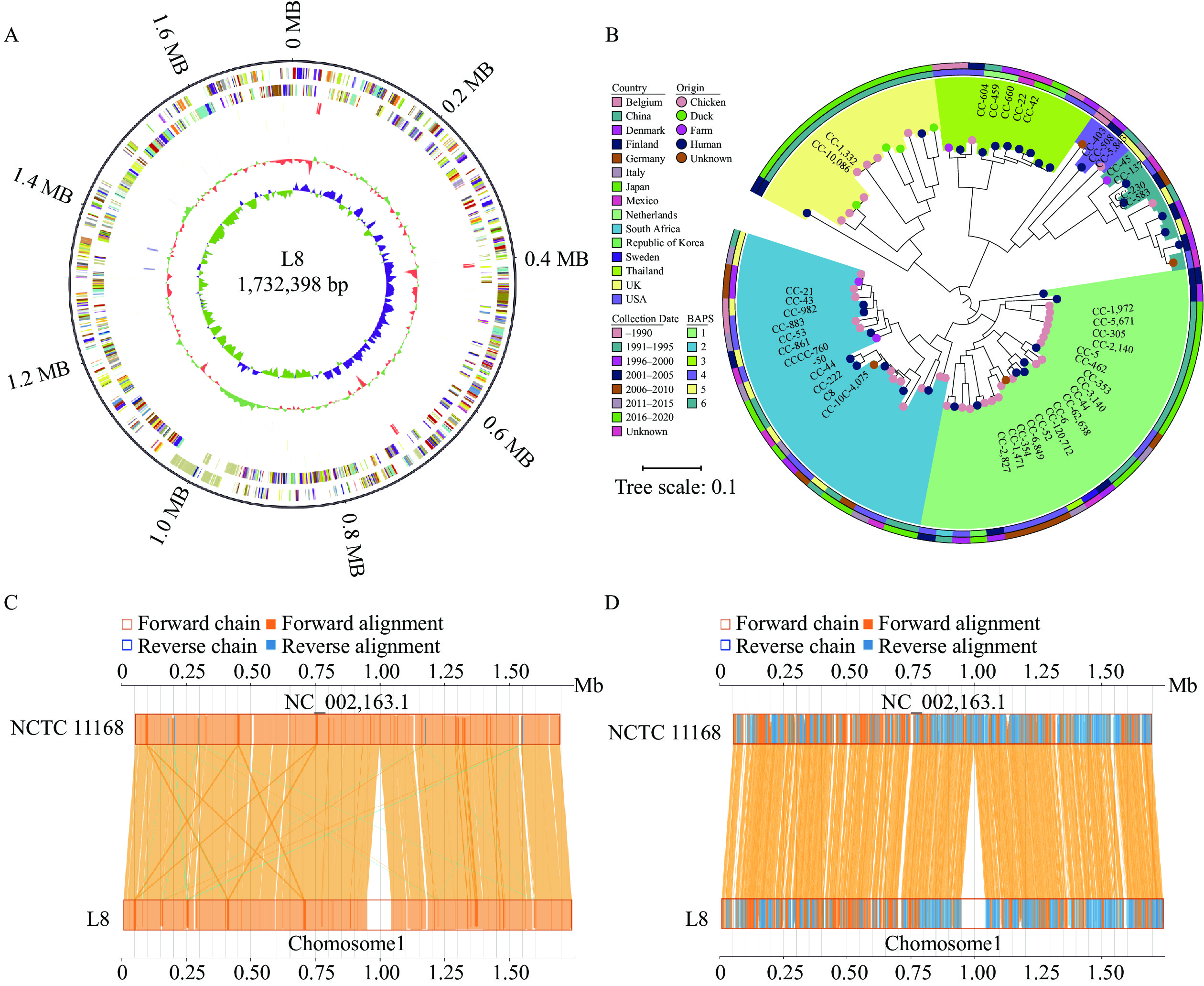 Figure 1.
Figure 1.Complete genomes of C. jejuni L8, comparative genomic analysis with NCTC 11168, and population structure of 84 C. jejuni strains based on the core genome alignment with BAPS clusters. (A) Circular representation of the chromosome from C. jejuni L8. (B) Population structure of 84 C. jejuni strains based on the core genome alignment with BAPS clusters.(C) Co-linear analysis of the genome between C. jejuni L8 and C. jejuni NCTC 11168 using MUMmer. (D) Co-linear analysis of the proteome between C. jejuni L8 and C. jejuni NCTC 11168 using MUMmer.
Note: In panel B, the clonal complexes are color-coded in the inner ring; the country of genome origin is coded in the second ring; and the collection date of the genome is described in the outer ring. The leaves are colored by the origin of each sample. In panel C, the yellow connecting lines in the middle region indicate high sequence identity of the forward alignment, and the blue lines represent the reverse complementary alignment.The 84 C. jejuni genomes, including L8, were isolated from poultry, human, and environmental samples from various countries. These genomes comprised 600 core genes, covering 54.6% of the average genome size of 1,694,827 bp. Phylogenetic analysis revealed six distinct branches (1–6), confirmed by BAPS clustering (Figure 1B). These branches were characterized by their clonal complexes (CCs), sequence type, geographic location, isolation source, and collection year (
Supplementary Table S2 ). L8 belonged to sequence type ST-464, assigned to BAPS cluster 1, which included isolates from eight countries. A total of 126 common virulence genes and 83 shared virulence factors were identified in L8, NCTC 11168, RM 1221, and 81-176, representing the fundamental elements contributing to C. jejuni virulence (Figure 2A). The distribution of virulence factors was most similar between L8 and NCTC 11168. L8 harbored 27 virulence genes, exhibiting the greatest diversity compared to 81-176 (15 virulence genes), excluding RM 1221, which contained the fewest virulence factors (Figure 2B). The previously identified virulence factor CDT was present in L8. Phylogenetic analysis of the cdtA gene indicated significant differences in L8 compared to other strains (Figure 3, cdtA), although no distinct variations were observed for cdtB and cdtC (Figure 3, cdtB, cdtC). Additionally, other virulence traits, such as lipooligosaccharide sialylation and the metabolism-related virulence factors γ-glutamyl transpeptidase (GGT) and fucose permease (Cj0486), were absent in L8.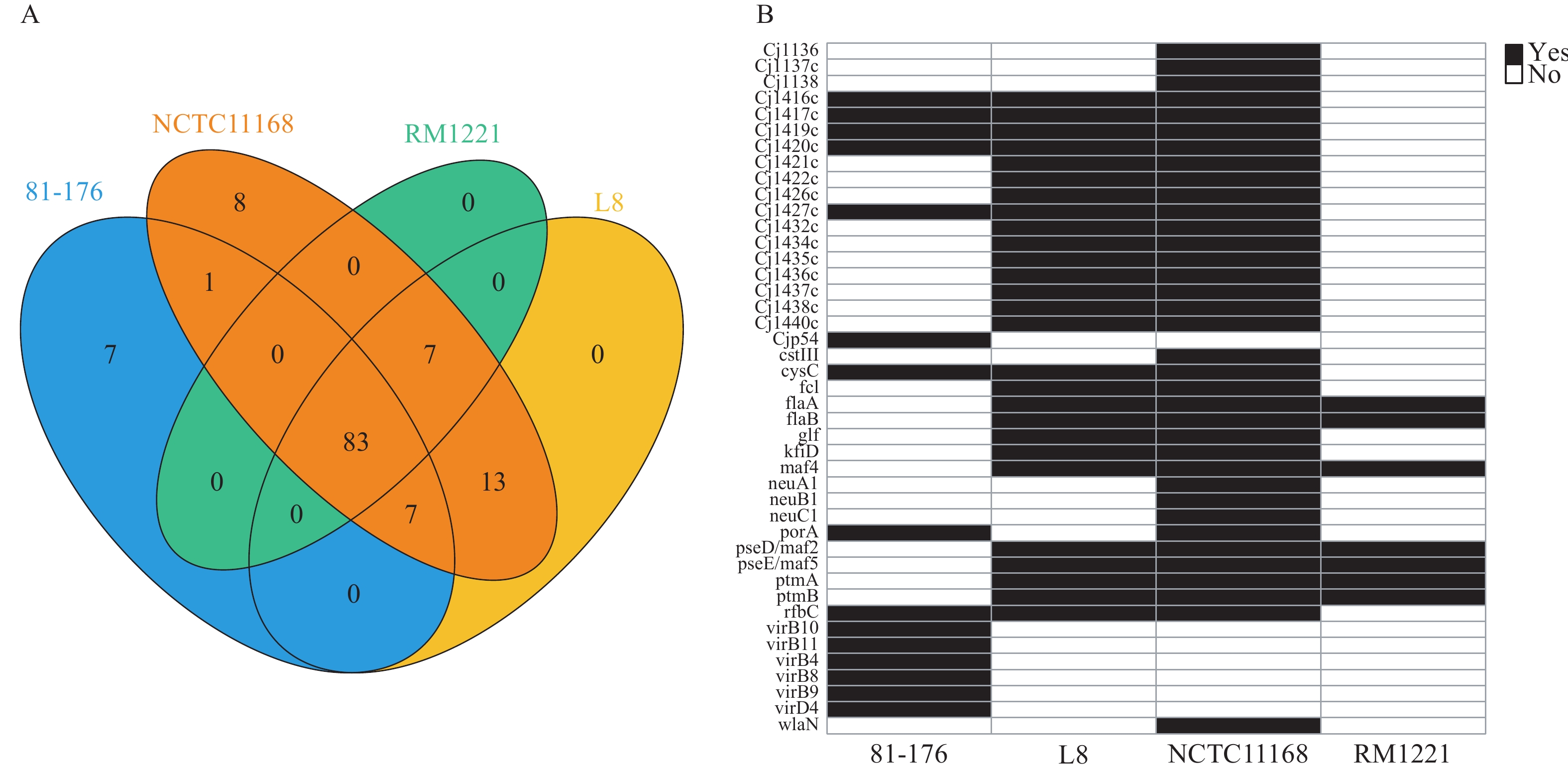 Figure 2.
Figure 2.Distribution comparison of 126 virulence genes in the four strains. (A) Venn diagram of the relationship between the 126 genes identified in the four strains. (B) Heat map of the distribution of the remaining 43 virulence factors except the 83 shared.
Note: A total of 98, 119, 90, and 110 genes were detected in C. jejuni 81-176, NCTC 11168, RM 1221, and L8, respectively. L8 shared 110 genes with NCTC 11168, 90 genes with RM 1221, and 90 genes with 81-176.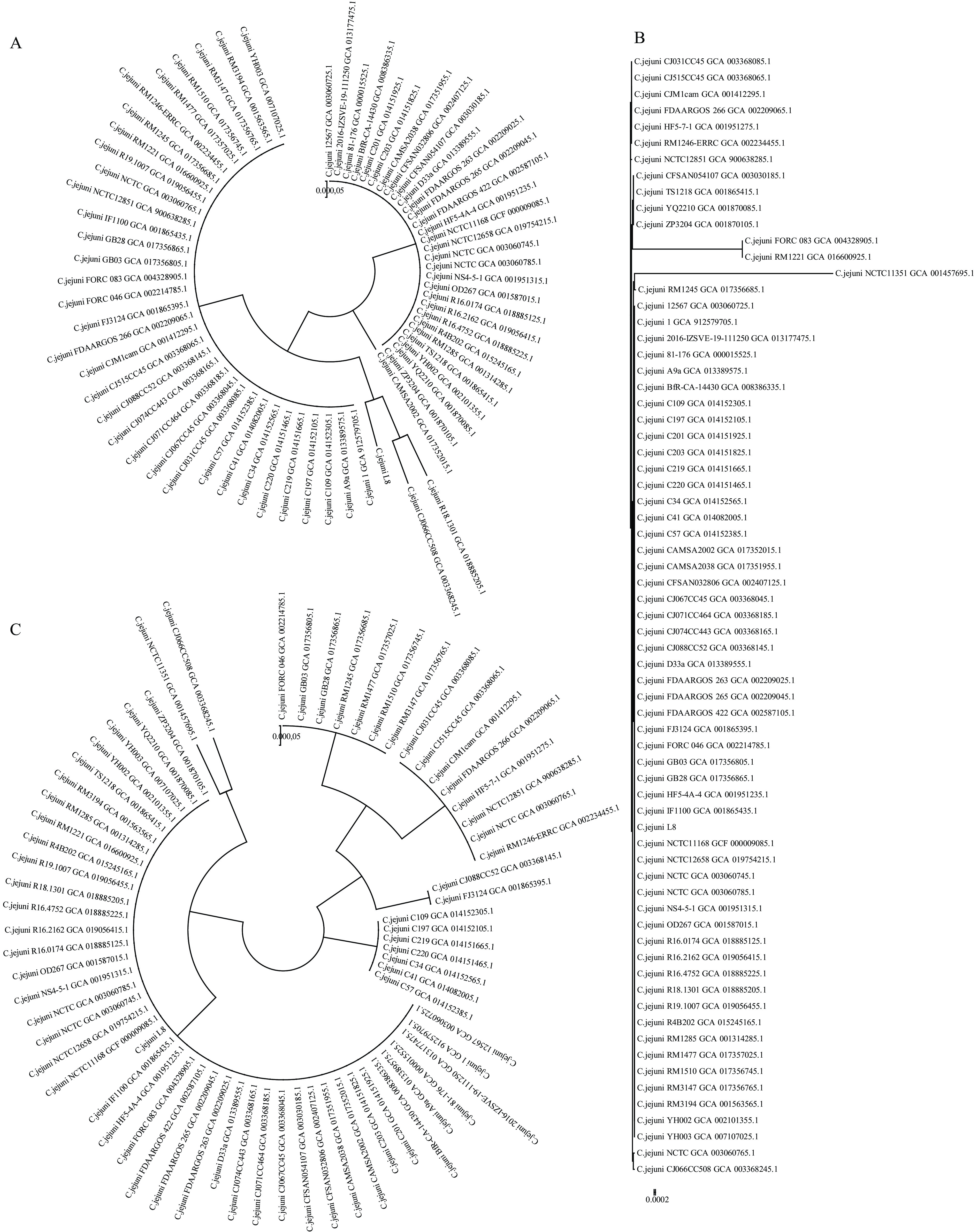 Figure 3.
Figure 3.Phylogenetic tree of predicted amino acid sequence variants encoded by (A) cdtA, (B) cdtB, and (C) cdtC in the 84 C. jejuni strains from different geographic backgrounds.
Note: CdtA was predicted in 67 of the 84 C. jejuni strains, and both CdtB and CdtC in 69. The L8 strain is denoted with a black triangle.Strain L8 harbored the 13 T6SS core components. Comparisons with T6SS-positive C. jejuni 108 and 488 revealed a strongly conserved T6SS cluster, sharing synteny in the genomic arrangement among C. jejuni strains (Figure 4A). In L8, the C-terminal tssI (vgrG) domain from positions 561 to 884 differed observably from those in 108 and 488. Notably, in L8, 25 amino acids were inserted at positions 566 to 590 and 16 amino acids at positions 596 to 611, respectively (Figure 4C). In contrast, the amino acid sequences at the same positions of 108 and 488 showed high consistency of 99.38%. Furthermore, L8 exhibited consecutive or scattered mutations outside of the two insertion segments from 561 to 884. Additionally, CJIE3 was identified in L8.
 Figure 4.
Figure 4.Comparison of 13 T6SS core components among L8, 108, and 488. (A) Organization of T6SS genes encoded by C. jejuni L8, 108, and 488. (B) Alignment of amino acid sequences of 11 genes. (C) Alignment of the amino acid sequence of TssI (VgrG) from the three C. jejuni strains after 560.
Note: The sequence alignment of TssI (VgrG) was truncated at 560. There was no change in the sequence alignment of TssB and TssE. Insertion sequences only in L8 are highlighted orange, and variant sequences are highlighted blue compared with the other two strains. -
Here, we isolated strain L8 from a clinically reported case of suspected tularemia and identified it as C. jejuni through biochemical characteristics and whole-genome sequencing. Genomic comparisons revealed the presence of a T6SS-containing CJIE3 in L8, considered a novel variant of the pathogenicity island (3). Notably, the insertion of two long segments and multiple consecutive or scattered mutations were present in L8’s C-terminal VgrG, a crucial effector of the conserved T6SS. In the search for specific virulence factors, we found that L8 contained the complete CDT gene, with CdtA showing significant differences from those in other strains.
In bacterial infections, a crucial virulence determinant is the T6SS, which forms a nano-crossbow-like structure in the attacker cell’s cytoplasm that propels an arrow composed of a haemolysin co-regulated protein tube and a VgrG spike to puncture the prey’s cell wall (4). VgrG is an essential and conserved structural component in all reported T6SSs to date (5–6), and the C terminus of VgrG is widely conserved and necessary for functional T6SS assembly (5). Surprisingly, L8’s C-terminal VgrG domain exhibited significant changes from positions 561 to 884, including the insertion of two long fragments and multiple consecutive or scattered mutations on their exterior. These alterations are very rare in previously reported T6SS structures (3). In contrast, the amino acid sequences between positions 108 and 488 showed 99.38% consistency. We hypothesize that this C-terminal VgrG domain was likely transformed into a toxin protein that may be associated with the patient’s clinical phenotype. Notably, this study did not elucidate the specific role of this unique structure in VgrG function. Nevertheless, this finding implies that this unusual VgrG structure may enhance the initial impact of T6SS on bacterial antagonism, subversion of host cells, and niche colonization, raising the possibility that the injection of toxin proteins into host cells led to high fever and convulsions in the child.
Comparative analysis of genes within the T6SS core components showed greater similarities between L8 and 108 for tagH, tssD, and tssA, while L8 and 488 were more similar for tssM, tssK, tssC, tssF, tssG, and tssI (<500). Multiple scattered single amino acid mutations identified within the T6SS core components of each strain indicate individual divergences. These findings suggest that these genes warrant further investigation to elucidate T6SS function.
Although L8 and NCTC 11168 exhibited high similarity in the distribution of common virulence genes, their clinical phenotypes differed significantly. Unfortunately, no specific virulence factors were identified in L8, unlike the potential virulence factors clearly observed in 81-176. However, phylogenetic analysis revealed that the subunit CdtA of CDT in L8 formed a distinct branch, indicating that its amino acid sequences were highly divergent from those of the other 66 strains. The toxic effects of CDT are primarily reflected in its ability to induce cell death and regulate the inflammatory response in human epithelial cells. C. jejuni CDT comprises three subunits: CdtA, CdtB, and CdtC. CdtA and CdtC bind to membrane lipid rafts, a crucial step for CdtB entry into cells (7). We hypothesize that the binding of CdtA variants and CdtC to membrane lipid rafts may facilitate CdtB entry into cells, potentially enhancing apoptosis and inflammation. Consequently, this series of processes likely exacerbated the patient’s clinical presentation, although further functional verification is required.
The sequence type of L8 was ST-464, assigned to BAPS cluster 1, where 25 of the 26 genomes originated from either chickens (17 genomes) or humans (8 genomes), with one exception isolated from a farm (Figure 1B). This suggests the patient was likely infected by a chicken, a conclusion supported by epidemiological investigation. The child had come into contact with chicken feces on his mouth due to thrush treatment using a folk remedy. The patient was initially admitted on acyclovir and then switched to cefoperazone/sulbactam a week later, and was discharged after another week. Interestingly, a similar case was reported in China, where a strain isolated from the blood of a child with bacteremia was initially identified as “Francisella” by the automatic bacterial identification instrument VITEK2.0, later confirmed as C. jejuni. Another case clinically suspected as tularemia was later identified as Paenibacillus assamensis (8). These instances underscore the unreliability of depending solely on automatic bacterial identification instruments for accurate strain identification. Additionally, a case of bloodstream infection by C. jejuni was reported in Guizhou Province, in which the patient presented with syncope and high fever without diarrhea (9).
This work might expand our understanding of the clinical manifestations of campylobacteriosis. Furthermore, we recommend considering the potential significance of T6SS in the pathogenesis of C. jejuni when studying the genetic characterization of clinical phenotypes (10).
-
No conflicts of interest.
-
the CDC workers who participated in the field epidemiological investigation and the doctors of Luxi County People’s Hospital who treated the patient.
HTML
| Citation: |

|