
 Download:
Download:




-
Nontyphoidal Salmonella enterica infections, particularly those caused by antimicrobial-resistant strains, are a major public health concern (1). Notably, fluoroquinolone (FQ)-resistant Salmonella spp. and extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae were listed by the World Health Organization in 2017 as high-priority pathogens posing the greatest threat to human health (2).
Resistance to these antimicrobials has been observed in S. enterica serotype Kentucky since the 2000s (3-5). Highly FQ-resistant S. Kentucky has become widespread in recent years (6), largely associated with the spread of ST198, which is the most common ST leading to human infections worldwide. Recently, a high prevalence of S. Kentucky ST198 with resistance to CIP and extended-spectrum cephalosporins (ESCs) was detected in human, environmental, and chicken samples in China (7-8).
In this study, we generated a genomic collection of 54 S. Kentucky ST198 strains recovered from an active surveillance system in Beijing over an 8-year period from 2016 to 2023. Our aim was to characterize the genetic features and antimicrobial resistance (AMR) profiles of S. Kentucky ST198 in Beijing.
-
Hospital-based active surveillance was conducted from 2010 onward in Beijing, China. From January 2016 to December 2023, a total of 41,742 diarrheal samples were collected from outpatients with gastroenteritis. Various food samples of animal origin were collected from retail outlets and supermarkets to screen for Salmonella spp. for food safety risk surveillance. All specimens were processed by routine microbiologic and biochemical tests to identify Salmonella. The Salmonella Kentucky serotype was detected by a slide agglutination test using commercially available antisera (SSI Diagnostica, Denmark) according to the White-Kauffmann-Le Minor scheme.
-
Antimicrobial susceptibility testing of Salmonella strains was conducted using the broth microdilution method following the guidelines of the Clinical and Laboratory Standards Institute document (CLSI M100-S29:2019). Twenty-eight antimicrobial agents belonging to 12 categories were tested: ampicillin (AMP), ampicillin-sulbactam (AMS), amoxicillin-clavulanic acid (AMC), cefazolin (CFZ), cefoxitin (CFX), cefotaxime (CTX), ceftazidime (CAZ), cefepime (FEP), kanamycin (KAN), gentamicin (GEN), streptomycin (STR), amikacin (AMI), tetracycline (TET), doxycycline (DOX), minocycline (MIN), nalidixic acid (NAL), ciprofloxacin (CIP), levofloxacin (LEV), gemifloxacin (GMI), sulfisoxazole (Sul), trimethoprim-sulfamethoxazole (SXT), chloramphenicol (CHL), azithromycin (AZI), aztreonam (AZM), colistin (CT), polymyxin B (PB), imipenem (IMP) and meropenem (MEM). An MDR phenotype was defined as resistant to at least three classes of antibiotics.
-
DNA was extracted using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Genomic DNA concentration was determined by agarose gel electrophoresis and fluorometric analysis (Qubit 2.0). WGS was conducted using an Illumina PE150 platform with 100× coverage (Novogene Technology Co., Ltd, Beijing, China). Raw sequencing data were quality-checked, trimmed, and assembled de novo into a draft genome sequence using SPAdes 3.13.
Core genome multi-locus sequence typing (cgMLST) was performed using the BacWGSTdb service. AMR genes were screened using the NCBI AMRFinderPlus tool 3.1.1b (https://ftp.ncbi.nlm.nih.gov/pathogen/Antimicrobial_resistance/AMRFinder/). Whole genome single nucleotide polymorphism (wgSNP) analysis for all draft genomes was conducted using snippy pipeline v.4.4.5 with the reference strain S. Kentucky ST198 PU131 (GenBank ID: CP026327). Single nucleotide polymorphism (SNP) distance matrices were obtained for all isolates using snp-dist v.0.6.3. The phylogenetic tree and heatmap of resistance genes were visualized using ChiPlot (https://www.chiplot.online/).
-
Between 2016 and 2023, an active surveillance system collected 1,838 S. enterica strains, of which 54 (2.9%) were S. Kentucky isolates. All 54 S. Kentucky isolates were assigned to ST198. Forty-nine were collected from patients with clinical diarrhea, and five were isolated from chicken meat. Eight and 23 of the 54 strains were from two outbreaks in 2016 and 2020, respectively, while the remaining isolates were from sporadic cases. These 54 strains were isolated between 2016 and 2023, with the following annual distribution: eight in 2016, two in 2018, two in 2019, 23 in 2020, three in 2021, seven in 2022, and nine in 2023. The strains were distributed across 12 districts.
-
All 54 S. Kentucky isolates were resistant to quinolones (NAL, CIP, LEV, and GMI), tetracyclines (TET and DOX), folate pathway inhibitors (Sul), and susceptible to carbapenems (IMP and MEM). Regarding cephems, S. Kentucky isolates showed the highest resistance to CFZ (90.7%), followed by CTX (87.0%), FEP (85.2%), and CAZ (63.0%). However, resistance to AMC, FOX, CT, and PB was relatively low, at only 1.9%. The frequency of AMR is presented in Figure 1. In addition, 85.2% (46/54) of ST198 isolates were ESBL-producing strains.
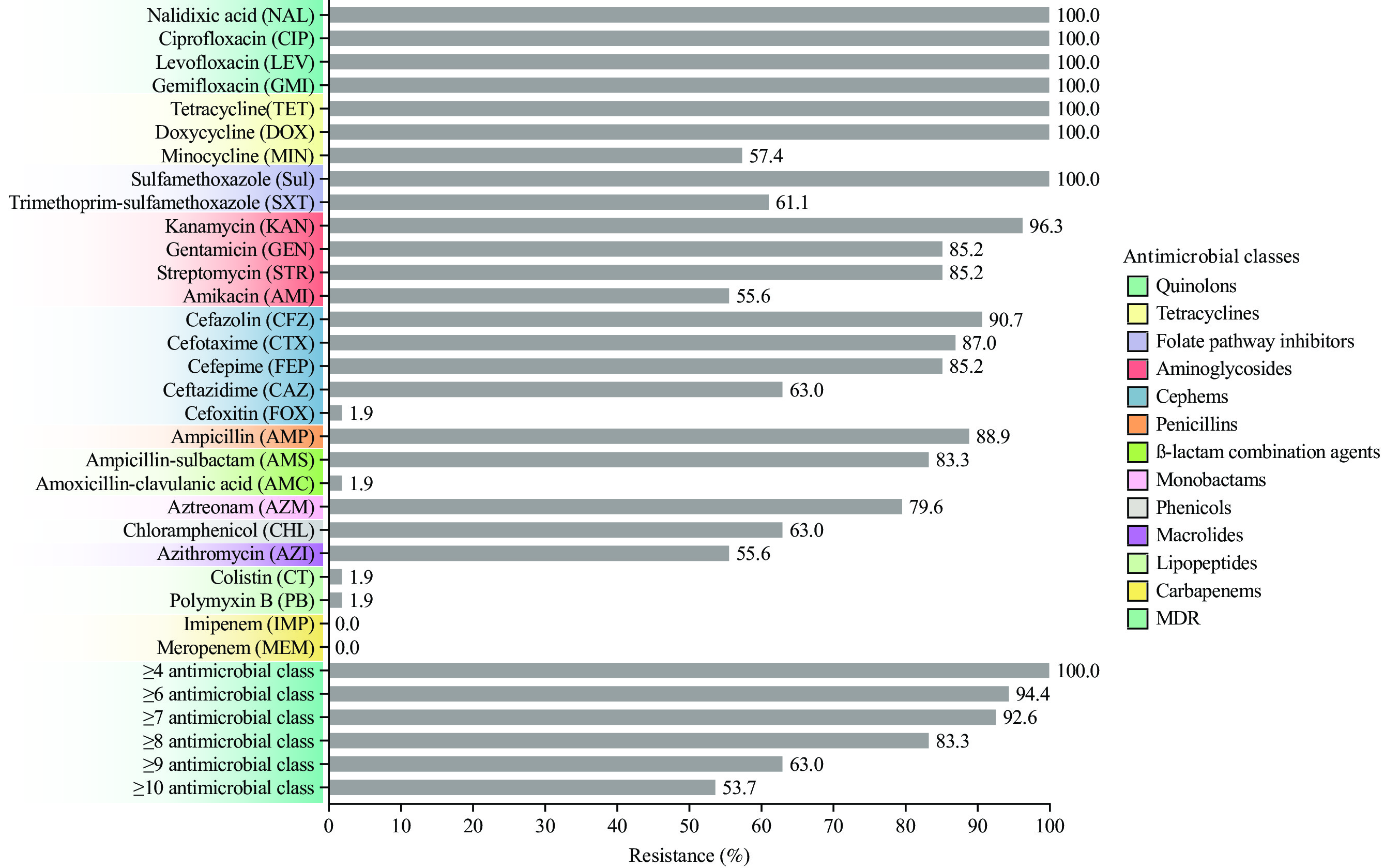 Figure 1.
Figure 1.Antimicrobial resistance of 54 S. Kentucky isolates against 28 antimicrobial agents belonging to 12 categories.
Abbreviation: S. Kentucky=Salmonella Kentucky; MDR=multidrug resistant.All 54 isolates (100%) were resistant to at least four antimicrobial classes. Resistance to six, eight, and 10 classes was observed in 94.4%, 83.3%, and 53.7% of isolates, respectively.
-
To reveal the genetic relatedness of S. Kentucky ST198 isolates, a global phylogenetic tree was constructed using 121 isolates, including 15 global strains, 52 Chinese isolates obtained from 11 other Provincial-level administrative divisions (PLADs), and the 54 isolates from this study (Figure 2). As previously reported (9), the S. Kentucky ST198 isolates were divided into two clades, 198.1 and 198.2. Clade 198.2 was further subdivided into two subclades, 198.2-1 and 198.2-2. The 15 global strains were mainly distributed in clades 198.1, 198.2-1, and 198.2-2. Although the Chinese isolates showed significant diversity, they were concentrated in clades 198.1 (3.3%, 4/121), 198.2-1 (24.0%, 29/121), and 198.2-2 (60.3%, 73/121). Furthermore, 30 subclade 198.2-1 isolates obtained from five provincial-level administrative divisions (PLADs), including Xichuan, Guangxi, Hunan, Jiangsu, and Zhejiang, were detected between 2013 and 2023, while 73 subclade 198.2-2 isolates obtained from nine PLADs (Anhui, Fujian, Guangdong, Guangxi, Guizhou, Jiangsu, Liaoning, Shandong, and Zhejiang) were collected from 2015 to 2022. Strains from various sources (animal, chicken, duck, environment, food, and human) clustered together, implying cross-transmission between hosts.
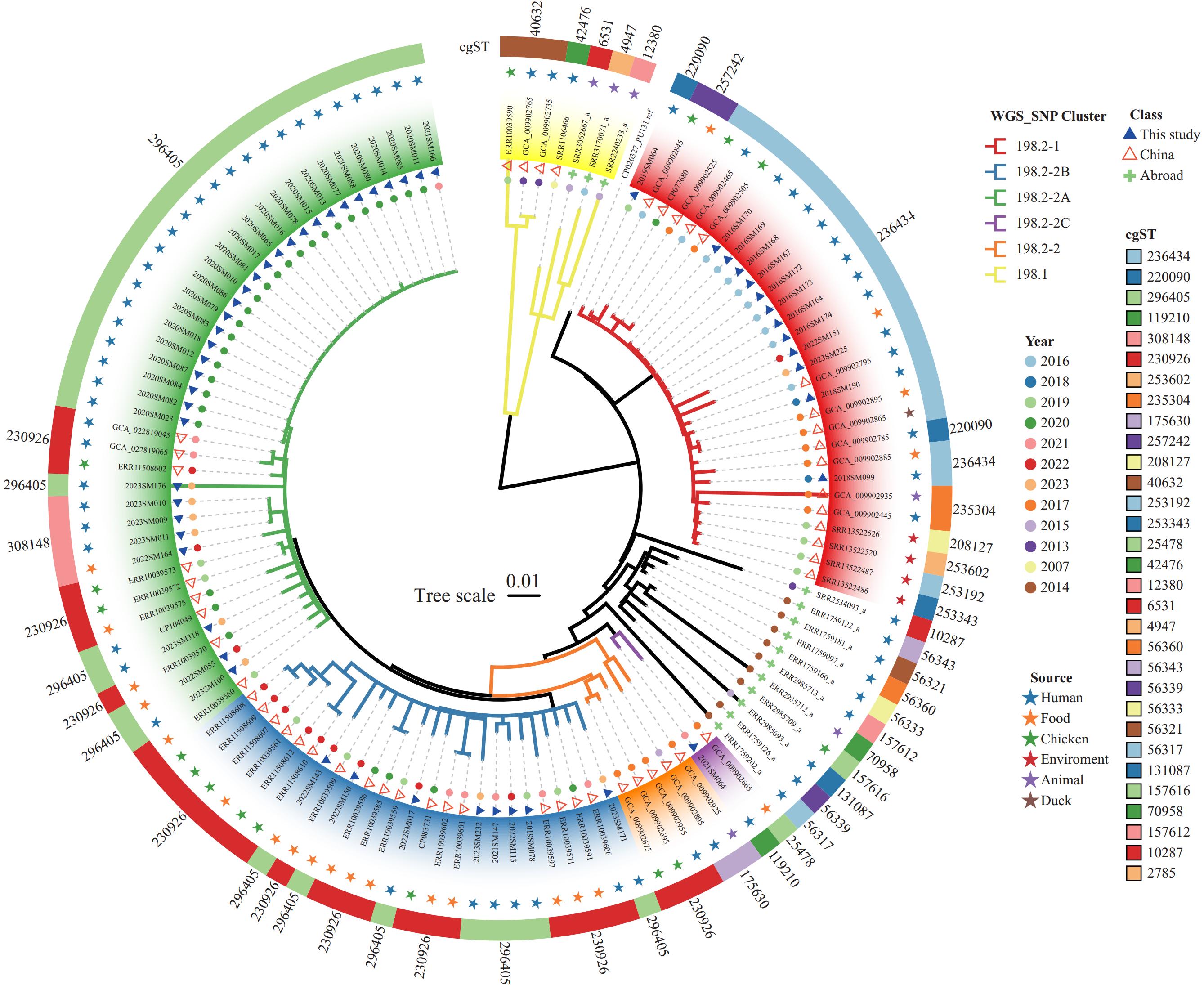 Figure 2.
Figure 2.The phylogenetic relationship of the 121 global S. Kentucky ST198 isolates based on wgSNPs.
Note: In the two outer circles, the cgSTs are shown in colored rings, and the sources of strains are shown in colored pentagrams. In the two inner circles, the isolation year of strains is indicated by colored dots, and strains from this study are shown as filled blue triangles, strains from China are shown as hollow red triangles, and global strains are shown as filled green crosses. The tree branches are color-coded to highlight S. Kentucky strains from clades 198.1, 198.2-1, 198.2-2, and sub-cluster 198.2-2 (2A-2C).
Abbreviation: S. Kentucky=Salmonella Kentucky; ST=sequence type; wgSNPs=whole genomic single nucleotide polymorphisms; cgST=core genomic sequence type.
To reveal the molecular characteristics of S. Kentucky ST198 isolates, we performed cgMLST on the whole-genome sequences of the 121 global isolates and identified 31 cgSTs (Figure 2). The most common cgSTs were cgST296405 (30.6%), followed by cgST230926 (23.1%) and cgST236434 (15.7%). Twenty-two cgSTs were represented by only one isolate, and cgST40632 was detected only in three clade 198.1 isolates. Among the 30 subclade 198.2-1 isolates, nine cgSTs were detected, with cgST236434 being the predominant type, accounting for 63.3% (19/30). By contrast, only six cgSTs were identified in 73 subclade 198.2-2 isolates, with cgST296405 accounting for 50.7% (37/73) and cgST230926 accounting for 38.4% (28/73).
-
Phylogenetic analysis of the 54 Beijing isolates was performed (Figure 3A), and a heatmap of SNPs was generated (Figure 3B). The number of SNPs ranged from 15 to 100 between food-derived isolates, from 0 to 103 between human-derived isolates, and from 9 to 102 between human- and food-derived isolates. Of the 54 Beijing isolates, 13 (24.1%), 32 (59.3%), 8 (14.8%), and 1 (1.9%) belonged to subclades 198.2-1, 198.2-2A, 198.2-2B, and 198.2-2C, respectively (Figure 3A). Thirteen isolates were distributed in subclade 198.2-1: one clustered with eight strains from outbreak 1 in 2016 (0 SNPs) in one branch, and the other clustered with five sporadic strains from 2018 and 2019 (1–4 SNPs). In subclade 198.2-2A, 23 strains from outbreak 2 in 2020 clustered together (0–1 SNP), along with nine sporadic strains, including seven human isolates from 2021 and 2023, and two food isolates (chicken meat) from 2022 (1–5 SNPs). In subclade 198.2-2B, the majority of isolates (n=8) clustered with six strains from humans (2019–2023) and two strains from food (2022) (1–4 SNPs). The remaining human isolate (2021SM064) in subclade 198.2-2C was distantly related to other Beijing isolates (22–31 SNPs).
 Figure 3.
Figure 3.Phylogenetic analysis and the AMR phenotypic and genotypic characteristics of the 54 S. Kentucky isolates from Beijing. (A) The phylogenetic relationships, epidemiological and molecular features, and the MDR phenotypes of the 54 S. Kentucky isolates. (B) The matrix of SNPs for the 54 S. Kentucky isolates shows all correlations between outbreak and sporadic strains, as well as the various sources. (C) A heatmap of AMR genes. The color blocks on the X-axis represent the categories of genes. Colored cells represent the presence of genes, and white cells represent the absence of genes.
Note: The size of the circle represents the number of SNP differences between two isolates: the larger the circle, the greater the number of SNP differences, and no circle indicates no difference (0 SNPs).
Abbreviation: AMR=antimicrobial resistance; S. Kentucky=Salmonella Kentucky; MDR=multidrug resistant; SNP=single nucleotide polymorphism.
The 54 Beijing isolates yielded five cgSTs: cgST296405 (66.7%), cgST236434 (22.2%), cgST308148 (7.4%), cgST220090 (1.9%), and cgST119210 (1.9%). The cgST296405 genotype was most prevalent, comprising 23 outbreak strains from 2020 and 13 sporadic strains from 2019 (n=1), 2021 (n=2), 2022 (n=5), and 2023 (n=5). The cgST236434 genotype included eight outbreak isolates from 2016 and four sporadic isolates from 2018 (n=2), 2022 (n=1), and 2023 (n=1). These findings were consistent with SNP analysis, suggesting that cgST296405 and cgST236434 represent two major endemic genotypes in Beijing.
-
To characterize the AMR profiles of the S. Kentucky ST198 isolates from Beijing, we scanned the genomes to identify AMR-related genes and mutations. We detected 32 AMR genes in 14 classes, including those involved in resistance to aminoglycosides (11 genes), β-lactams (3 genes), quinolones (1 gene and 4 point mutations), efflux pumps (2 genes), sulfonamides (2 genes), rifamycins (1 gene), trimethoprims (1 gene), tetracycline (1 gene), phenicols (1 gene), fosfomycin (1 gene), macrolides (1 gene), lincosamides (1 gene), colistins (1 gene), and quaternary ammonium (1 gene) (Figure 3C). Resistance genes from seven classes were found in strains within clade 198.2-1, while 14 classes were found in strains within clade 198.2-2.
The AMR gene profiles and phenotypes differed between the subclade 198.2-1 and 198.2-2 strains (Figure 3A and 3C). For β-lactamase resistance genes, blaCTX-M-14b was the only gene detected among the 13 subclade 198.2-1 isolates. However, in subclade 198.2-2, blaCTX-M-55 and blaTEM-1B were detected in 82.9% (34/41) and 73.2% (30/41) of isolates, respectively. For the aminoglycoside resistance gene profiles, aac(3)-Id, aadA7, and aph(3')-Ia genes were detected in 98.1% (53/54) of isolates, and the aph(3'')-Ib gene was detected in 25.9% (14/54) of isolates. The aac(3)-IId, aadA2, and rmtB genes were only detected in subclade 198.2-2, accounting for 78.0% (32/41), 75.6% (31/41), and 70.7% (29/41) of isolates, respectively. The aph(3'')-Ib and aph(6)-Id genes were detected in all 41 subclade 198.2-1 isolates but only one subclade 198.2-2 isolate. For tetracycline resistance genes, tet(A) was detected in all 198.2-1 isolates and 97.6% (40/41) of 198.2-2 isolates. For folate pathway antagonist genes, sul1 was prevalent in all isolates, and sul2 was only detected in two 198.2-2 isolates. The dfrA14 and floR genes were detected in 78.0% (32/41) of the 198.2-2 isolates. Additionally, all strains resistant to SXT carried at least one gene associated with resistance to folate pathway antagonists. The genes lnu(F), mph(A), arr-2, and fosA3, conferring correspondent resistance to lincosamide, macrolide, rifamycin, and fosfomycin, were only detected in 198.2-2, accounting for 75.6% (31/41), 68.3% (28/41), 78.0% (32/41), and 78.0% of isolates, respectively. For quinolone resistance, the same mutations in parC_S80I and gyrA_S83F were detected in all 198.2 isolates, but gyrA_D87G was present in 198.2-1 isolates, whereas gyrA_D87N was present in 198.2-2 isolates. The qnrS1 gene was only detected in 58.5% (24/41) of 198.2-2 isolates. Notably, the mcr-1 gene was detected in only one strain, 2022SM055 from chicken meat, which was found to be resistant to colistin. Importantly, the 198.2-2 isolates showed more complex AMR phenotypes and carried more resistance genes than the 198.2-1 isolates.
-
To demonstrate the evolutionary relationships of S. Kentucky strains from different sources in Beijing, a Sankey diagram was constructed (Figure 4). The first outbreak, occurring in Yanqing District in 2016, involved eight human-derived strains belonging to phylogenetic lineage 198.2-1 and cgST236434 (Figure 2, Figure 4). The second outbreak, occurring in four districts (Xicheng, Yanqing, Haidian, and Huairou) in 2020, involved 23 human-derived strains grouped in lineage 198.2-2A and cgST296405, suggesting cross-regional transmission and clonal expansion during this outbreak. The remaining 23 sporadic cases, distributed across 11 districts (Daxing, Docheng, Fangshan, Fengtai, Haidian, Huairou, Mentougou, Pinggu, Shunyi, Tongzhou, and Xicheng) between 2018 and 2023, included 18 human-derived and five chicken meat-derived strains. These sporadic strains derived from a polyclonal evolutionary source (198.2-1, 198.2-2A, 198.2-2B, and 198.2-2C) and formed five cgMLST groups. The five chicken meat isolates, distributed across two districts in 2022, were mainly from lineages 198.2-1, 198.2-2A, and 198.2-2B and were closely related to human isolates. These results suggest that the S. Kentucky ST198 outbreak isolates have two predominant clonal sources: 198.2-1 with cgST236434 before 2019 and 198.2-2A with cgST296405 after 2019.
 Figure 4.
Figure 4.A Sankey diagram demonstrating the spatiotemporal distribution and the transmission of multiple lineages of S. Kentucky strains in Beijing.
Abbreviation: S. Kentucky=Salmonella Kentucky. -
S. Kentucky ST198 has been increasingly reported in chickens and can cause human infections in China (8–9). In this study, all 54 S. Kentucky isolates were assigned to ST198. Our findings showed that the positive ratio for S. Kentucky ST198 among S. enterica isolates was 2.9% (54/1,838) in Beijing, which is approximately fourfold higher than that reported in a previous study (0.39%, 40/16,247) (9) and much higher than that reported in Shenzhen (0.7%, 57/8,559) (10) and among patients (0.33%, 40/12,011) in China between 2010 and 2016 (8).
In the present study, genomic analyses (cgMLST and wgSNP) were conducted to characterize the S. Kentucky ST198 strains. Five cgSTs were identified, with cgST296405 and cgST236434 being the predominant genotypes causing outbreaks. Phylogenetic analysis indicated that the 54 S. Kentucky strains clustered into lineages 198.2-1 and 198.2-2, then further divided into three sublineages, 198.2-2A, 198.2-2B, and 198.2-2C, revealing the existence of multiple lineages circulating in Beijing. Additionally, many sublineages were shared by strains from this study and strains from other PLADs, such as Guangxi, Shandong, Fujian, and Zhejiang, indicating a potential phylogenetic relationship between these strains.
In view of the increasing public health threat posed by the global emergence and dissemination of antimicrobial-resistant S. Kentucky, this investigation revealed a high level of both phenotypic and genotypic resistance to different classes of antimicrobials among 54 S. Kentucky isolates. The isolates were resistant to ESCs as follows: CTX (87.0%, 47/54), CAZ (63.0%, 34/54), and FEP (85.2%, 46/54). Resistance to ESCs is normally mediated by the production of ESBLs, and CTX-M-type ESBLs pose a particularly serious public health threat worldwide (11). In this study, all Beijing isolates in subclade 198.2-1 carried the gene blaCTX-M-14b, consistent with previous studies in China (6–7,9,12), indicating the formation of a clade carrying blaCTX-M-14b in China. In addition, compared with blaCTX-M-14b in 198.2-1, the co-existence of blaCTX-M-55 and blaTEM-1B was detected in the majority of 198.2-2 isolates. It is worth mentioning that all S. Kentucky isolates in this study were resistant not only to CIP but also to other FQ antibiotics (LEV, GMI), showing substantially higher levels of resistance than previously reported for S. Kentucky in China or many other countries (6,8,13). Moreover, all strains carried two quinolone resistance-determining region mutations (gyrA_S83F and parC_S80I), but qnrS1 and gyrA_D87N were only detected in 198.2-2 isolates. Several studies reported the presence of the qnrS1 gene on the bacterial chromosome, along with a large number of chromosomally-located resistance genes, after 2017. The emergence of qnrS1 in S. Kentucky isolates with gyrA and parC mutations increased bacterial resistance to CIP (10,12). In the present study, up to 85.2% of isolates were both FQ-resistant and ESBL-producing, confirming these as common features of S. Kentucky. Our study further revealed that the acquisition of an MDR phenotype by 198.2-2 has potentially contributed to its higher prevalence compared with 198.2-1, making it the most prevalent subclade for clonal transmission in Beijing.
In conclusion, the human salmonellosis epidemic caused by S. Kentucky involved multiple native circulating lineages that have become widely distributed across Beijing districts since 2016. Many clinical isolates were genetically clonal to strains isolated from food, suggesting possible cross-host transmission. Increased genome sequencing of strains from various host sources will facilitate the identification of transmission routes and determine potential ongoing outbreaks, which is vital for formulating targeted surveillance and countermeasures.
-
No conflicts of interest.
HTML
Sample Collection and Salmonella Identification
Antimicrobial Susceptibility Testing
Whole-Genome Sequencing (WGS) and Genomic Analysis
Epidemiological Information for S. Kentucky ST198 Isolates from Beijing
Antibiotic Resistance and MDR Profiles
Phylogenetic Analysis of Global S. Kentucky ST198 Isolates
Diversity and Discrepancy of S. Kentucky ST198 Isolates from Beijing
Resistomes of S. Kentucky ST198 Isolates from Beijing
Spatiotemporal Distribution and Phylogenetic Relationship of Sporadic and Outbreak S. Kentucky Strains in Beijing
| Citation: |

|