

 Download:
Download:




-
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a single-stranded positive RNA virus, exhibits a high rate of genome mutation and recombination (1–2). Recombinant strains of this virus could potentially affect their binding ability to the ACE2 receptor, which may in turn diminish the protection offered by vaccines and neutralizing antibodies. Notably, the Delta and Omicron recombinations have been collectively referred to as Deltacron (3). Furthermore, XBB.1.5, a recombinant strain resulting from the combination of the BA.2 sublineages BA.2.10.1 and BA.2.75, has demonstrated enhanced transmissibility (4). As such, it is imperative to monitor the genome recombination of SARS-CoV-2 in order to provide valuable insights regarding epidemic and transmission trends.
In late 2022 and early 2023, SARS-CoV-2 mutation monitoring results from China revealed that the dominant lineages included BF.7.14, BA.5.2.48, and BA.5.2.49, among others (5). Importantly, the Chongqing CDC recently documented a case of co-infection involving both BF.7.14 and BA.5.2.48 (6). Based on this evidence, we hypothesize that genomic recombination may occur between the prevalent SARS-CoV-2 strains BF.7.14 and BA.5.2 in China.
We analyzed the SARS-CoV-2 genomic sequences submitted to GISAID from China (7) and identified nine potential recombinant sequences from Henan Province (n=2), Anhui Province (n=3), and Sichuan Province (n=4) (GISAID: EPI_SET_230323sq). A phylogenetic tree was constructed including early sequences from the BA.2, BA.5, BA.5.2, BA.5.2.6, BA.5.2.48, BA.5.2.49, BF.7, and BF.7.14 lineages. These nine recombinant sequences were classified within the BA.5.2.1 (abbreviated as BF) clade and exhibited the closest relationship with the BF.7 and BF.7.14 lineages (Figure 1A). Notably, the topology between these recombinant sequences and the BF.7 lineage was found to be complex and poorly supported.
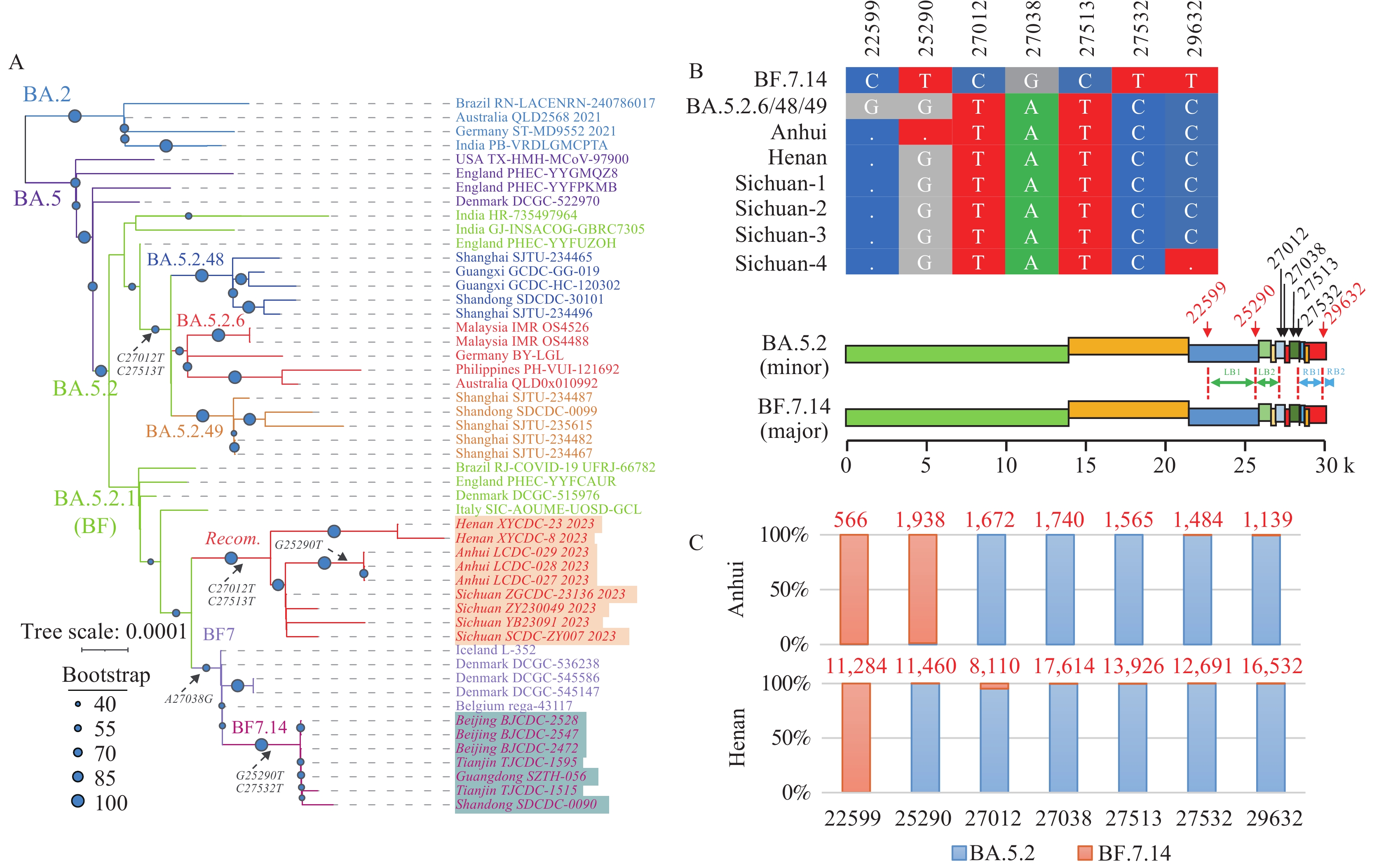 Figure 1.
Figure 1.Genome recombination between SARS-CoV-2 BF.7.14 and BA.5.2 sublineages. (A) Phylogenetic tree of the recombined SARS-CoV-2 genomes; (B) The nucleotides of seven key sites; (C) Validation of the mutations of key sites using original sequencing datasets.
Note: In panel (A), the key mutations and sequences were italicized. In panel (B), positions with mutations used to determine the break points were colored in red. In panel (C), sequencing depth of the key sites were colored in red.
Abbreviation: SARS-CoV-2=severe acute respiratory syndrome coronavirus 2; LB=left breakpoint; RB=right breakpoint.
Relative to the BF.7.14 lineage, all recombinant sequences carried two defining mutations (C27012T and C27513T) of the BA.5.2 lineage. In addition, the A27038G mutation, a defining feature of the BF.7.14 lineage, was absent from these sequences. Consequently, we inferred that these nine sequences from the three provinces may have resulted from genomic recombination between the BF.7.14 and BA.5.2 lineages (Figure 1B). Moreover, we examined the original sequencing datasets of these sequences from Henan and Anhui provinces and validated the key mutation sites within the recombination region (Figure 1C).
The identified recombinant sequences primarily consisted of the BF.7.14 framework, with a relatively low contribution from the BA.5.2 lineage. All nine recombinant sequences contained the signature mutation G22599C (S:R346T) found in the BF.7 lineage. Six of the sequences, not including the three from Anhui Province, were aligned with the BA.5.2 lineage at position 25290. We suspected that the left breakpoint of these recombination events could be situated between positions 22599 and 25290, and designated this region as Left Breakpoint 1 (LB1). The G25290T mutation in the three Anhui Province sequences matched that of BF.7.14. Considering the low likelihood of multiple SARS-CoV-2 recombination events occurring within a brief period, we hypothesize that the left breakpoint of the recombination event in the Anhui sequences might be located between positions 25290 and 27012 (LB2). Intriguingly, a sequence from Sichuan Province (Sichuan-4, EPI_ISL_16737882) shared the C29632T mutation with BF.7.14, suggesting that the right breakpoint of the recombination event in this particular sequence might lie between positions 27532 and 29632, which we refer to as Right Breakpoint 1 (RB1). The remaining sequences were consistent with the BA.5.2 lineage at position 29632, implying that the recombination fragment might extend towards the end of the sequences. Consequently, we provisionally define the region between 29632 and the genome’s three prime ends.
The primary recombination regions are situated from the tail of the Spike gene to the 3’ end of the genome. The S protein of the recombinant sequence, particularly the S1 portion containing the receptor binding domain (RBD), retains the majority of the BF.7.14 characteristics. Consequently, we cautiously hypothesize that the effect of this recombination event on the existing transmission and entry routes of the coronavirus in China may be limited. Given the relatively low number of reported cases involving the recombinant virus, additional research is required to ascertain whether it will result in severe clinical symptoms in infected individuals.
-
Sichuan Provincial Center for Disease Control and Prevention for uploading the sequencing data to GISAID.
HTML
| Citation: |

|