
 Download:
Download:




-
Vibrio parahaemolyticus (V. parahaemolyticus) is a halophilic bacterium that is naturally present in marine and estuarine environments and responsible for acute diarrheal illness in humans (1). In many Asian countries, such as China and Japan, V. parahaemolyticus is becoming one of the leading causes of food-borne infections (2).
The thermolabile hemolysin gene (tlh) is considered a signature molecular marker for V. parahaemolyticus (1). The virulence-associated genes of the species are thermostable direct hemolysin gene (tdh) and TDH-related hemolysin gene (trh). Up to 90% of clinical isolates of V. parahaemolyticus possess tdh and/or trh genes (1,3). Since 1996, O3:K6 serotype of V. parahaemolyticus has been prevalent in the world and causing pandemics (1,4). The pandemic clone has characteristics of tdh+ trh- toxRS/new+ (a unique toxRS sequence) orf8± (the orf8 sequence of f237 phage) (5–6). The Type III secretion system (T3SS), including gene clusters T3SS1 and T3SS2, has also been shown to be associated with the pathogenicity of V. parahaemolyticus, which is involved in cytotoxicity to host cells and related to the enterotoxicity of V. parahaemolyticus (1,7).
We previously reported the serology and antimicrobial susceptibility of clinical V. parahaemolyticus strains, among which the O3:K6 has been the consistently dominant serotype in Beijing from 2010 to 2019 (8). However, an emerging serotype O10:K4 from clinical isolates was identified and became the most prevalent locally in 2021. The molecular characteristics which contributed to the survival and spread of this particular clone are rarely known. Therefore, whole genome sequence-based analysis of these isolates is of utmost importance to elucidate their genetic characteristics, pathogenicity and transmission.
-
Hospital-based active surveillance has been conducted since 2010 in Beijing, China. The sentinel hospitals affiliated with 16 different districts enrolled outpatients with acute diarrhea. The average monthly enrollment number was around 20–40 patients per district. A total of 5,337 cases were collected from January to December 2021. Enrollment was subject to obtaining informed verbal consent. All specimens were collected on the day of presentation by rectal swabs in Cary-Blair transport media and were immediately transported to the laboratory of the District Center for Disease Prevention and Control (CDC) for processing within 24 hours.
-
For selective enrichment of Vibrio spp., swabs were inoculated on peptone water containing 3% NaCl, pH 8, incubated at 37 °C overnight, then inoculated on CHROMagar Vibrio media (CHROMagar Co., Paris, France), and incubated for 16–24 h. After culturing, at least three suspected colonies were picked out for further identification. The systematic identification was confirmed with the VITEK 2 Compact instrument (bioMérieux, Marcyl’Etoile, France). Finally, serologic identification was performed by a slide agglutination test with 11 O (lipopolysaccharide) and 65 K (capsule) antisera (Denka Seiken Ltd., Tokyo, Japan). One serotype was defined as a unique combination of O and K serogroups.
-
Antimicrobial susceptibility testing (AST) of V. parahaemolyticus strains was assessed using the broth microdilution method following the guidelines of the Clinical and Laboratory Standards Institute document (CLSI M100-S29:2019). Escherichia coli ATCC 25922 was included in the test as a quality control strain. Seventeen antimicrobial agents (Shanghai Xingbai Co) were used for AST: chloramphenicol, trimethoprim-sulfamethoxazole, colistin, ertapenem, meropenem, cefotaxime, ceftazidime, ceftazidime-avibactam, tetracycline, tigecycline, ciprofloxacin, nalidixic acid, aztreonam, amikacin, streptomycin, ampicillin, and ampicillin-sulbactam.
-
DNA was extracted using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). Quantification of extracted genomic DNA (gDNA) was determined by agarose gel electrophoresis and fluorometric analysis (Qubit 2.0). Whole genome sequencing (WGS) was conducted using an Illumina PE150 platform with 200× coverage (Novogene Technology Co., Ltd., Beijing, China). Raw sequencing data was checked for quality, trimmed, and assembled de novo into contigs. Whole genome sequencing-single nucleotide polymorphism (WGS-SNP) analysis for all draft genomes was performed using parsnp software with the reference strain sequence GCF_000196095.1 available from NCBI’s genome database. The phylogenetic tree was finally visualized using the online tool iTOL (
http://itol.embl.de/ ). -
The genomic analysis was based on the Center for Genomic Epidemiology’s web server (
https://cge.cbs.dtu.dk/services/cge/ ). Multilocus sequence typing (MLST) 2.0 was performed using seven housekeeping genes (dnaE, gyrB, recA, dtdS, pntA, pyrC, and tnaA) to characterize sequence type (ST) of V. parahaemolyticus isolates. The new STs were submitted to PubMLST (tted to PubMLST (https://pubmlst.org/organisms/vibrio-parahaemolyticus ). ResFinder 4.1 was used for screening antimicrobial resistant genes (ARGs). The virulence-associated genes (VGs) were found using virulence factor database (VFDB) (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi ). -
73 out of 5,337 (1.4%) diarrheal outpatients were positive for V. parahaemolyticus in 2021. Serological analysis of the 73 V. parahaemolyticus isolates revealed a total of 7 serovars with 3 defined serotypes (O10:K4, O3:K6, and O6:K18) and 4 kinds of untypeable K antigens. O10:K4 (83.6%, 61/73) was the most common one, followed by O2:KUT (5.4%, 4/73), O4:KUT (4.1%, 3/73), O1:KUT (2.7%, 2/73) and O3:K6, O6:18, and O10:KUT each (1.4%, 1/73) (Table 1). These results indicated the emerging serotype O10:K4 had replaced O3:K6, which accounted for 67.7% of clinical isolates during the period of 2010–2019 (8), becoming the predominant serotype in 2021.
Serovars No. of isolate (s) ST Virulence genes Pandemic markers tdh trh toxRS/new orf8 O10:K4 61 ST3 + - + + O3:K6 1 ST3 - - + - O6:K18 1 ST1490 - - + - O1:KUT 1 ST3 + - + - 1 ST2620 - - + - O2:KUT 4 ST2781, ST2894, ST2895, ST2896 - - + - O4:KUT 1 ST499 - - + - 2 ST2516 + - + - O10:KUT 1 ST2897 - - + - Abbreviations: ST=sequence type; V. parahaemolyticus=Vibrio parahaemolyticus. Table 1. Serotypes, ST and virulence factors of 73 clinical V. parahaemolyticus strains in Beijing, 2021.
-
The antimicrobial susceptibilities of 73 V. parahaemolyticus strains were listed in Table 2. All isolates were sensitive to the following 14 antimicrobials agents such as ampicillin-sulbactam, ceftazidime-avibactam, cefotaxime, ceftazidime, ertapenem, meropenem, amikacin, tetracycline, aztreonam, tigecycline, nalidixic acid, ciprofloxacin, trimethoprim-sulfamethoxazole and chloramphenicol. Only 2.7% of 73 isolates were resistant to colistin and 97.3% demonstrated intermediate resistance to colistin. Additionally, the sensitivity rates of 73 isolates to ampicillin and streptomycin were 89.0% and 67.1%, respectively, and the intermediate resistance rates were 11.0% and 32.9%, respectively. The ARGs analysis showed that all 73 strains carried at least one of the 9 kinds of β lactamase resistance genes (blaCARB-18, blaCARB-20, blaCARB-22, blaCARB-24, blaCARB-29, blaCARB-30, blaCARB-33, blaCARB-34, and blaCARB46) (Figure 1). Two strains had the quinolone resistance gene qnrC. Interestingly, all 61 O10:K4 strains carried blaCARB-22.
Antimicrobial class Antimicrobial agent Susceptible
n (%)Intermediate
n (%)Resistant
n (%)Penicillins ampicillin 65(89.0) 8(11.0) 0 ß-Lactam/ß-lactamase ampicillin-sulbactam 73(100.0) 0 0 inhibitor combinations ceftazidime-avibactam 73(100.0) 0 0 Cephems cefotaxime 73(100.0) 0 0 ceftazidime 73(100.0) 0 0 Carbapenems ertapenem 73(100.0) 0 0 meropenem 73(100.0) 0 0 Aminoglycosides amikacin 73(100.0) 0 0 streptomycin 49(67.1) 24(32.9) 0 Macrolides aztreonam 73(100.0) 0 0 Tetracyclines tetracycline 73(100.0) 0 0 tigecycline 73(100.0) 0 0 Quinolons and fluoroquinolones nalidixic acid 73(100.0) 0 0 ciprofloxacin 73(100.0) 0 0 Folate pathway inhibitors trimethoprim-sulfamethoxazole 73(100.0) 0 0 Phenicols chloramphenicol 73(100.0) 0 0 Lipopeptides colistin 0 71(97.3) 2(2.7) Abbreviation: V. parahaemolyticus=Vibrio parahaemolyticus. Table 2. Antimicrobial susceptibility of 73 clinical V. parahaemolyticus strains in Beijing in 2021.
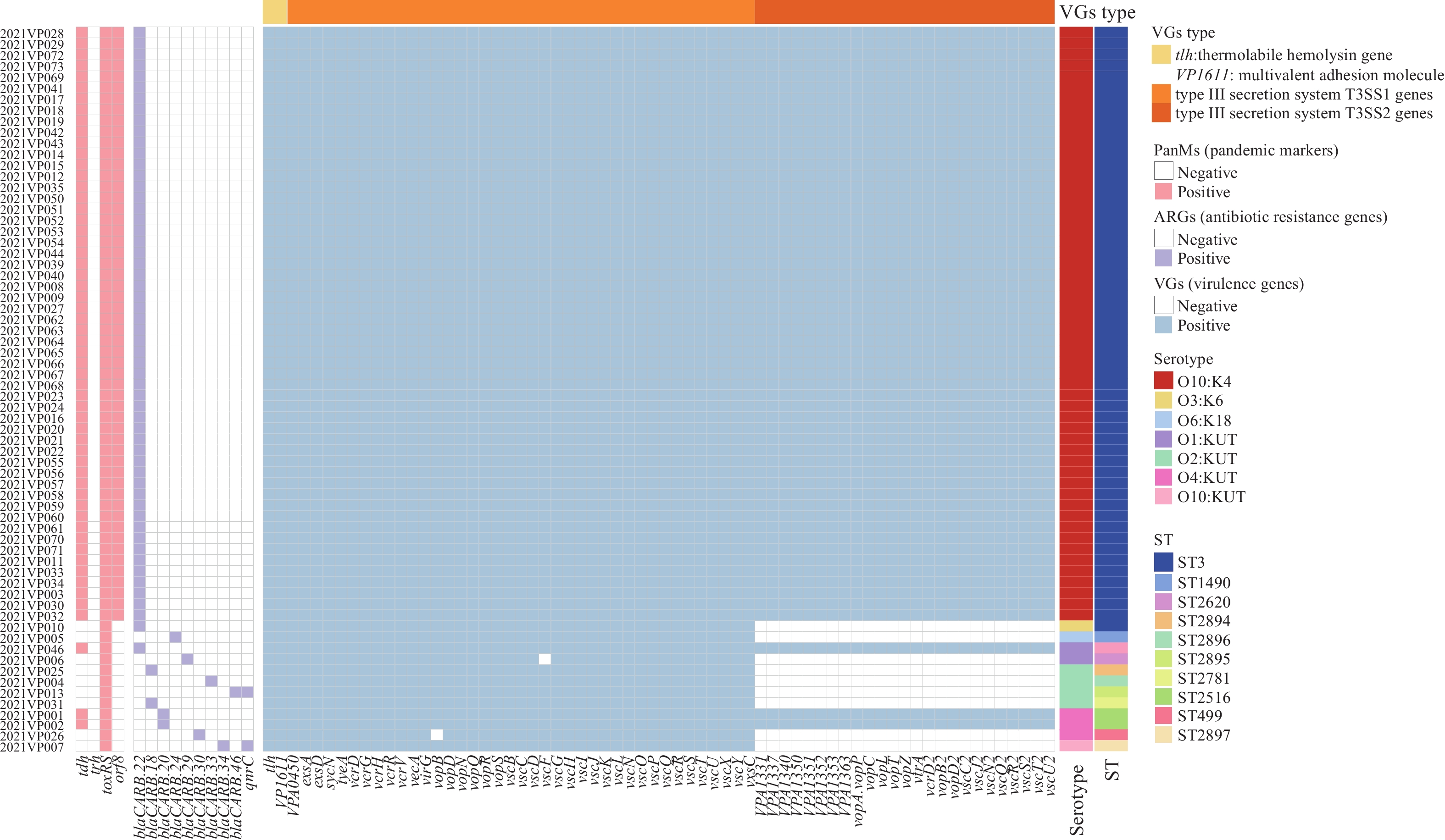 Figure 1.
Figure 1.Distributions of serotype, STs, antibiotic resistance genes, virulence genes, and pandemic markers among 73 clinical V. parahaemolyticus strains in Beijing in 2021.
Note: The color strips indicate areas corresponding to the isolates. Pink colored cells represent the presence of pandemic makers and white cells represent the absence of the pandemic markers; Lilac colored cells represent the presence of antibiotic resistance genes and white cells represent the absence of the antibiotic resistance genes; Light blue cells represent the presence of virulence-associated genes and white cells represent the absence of the virulence-associated genes. Abbreviations: ST=sequence type; V. parahaemolyticus=Vibrio parahaemolyticus. -
All of the 73 strains had the tlh gene, but none had the trh gene (Figure 1). 64 isolates (87.7%) were positive for the tdh gene, of which 61 strains carried the orf8 gene. The serotypes of these 64 tdh+ strains included O10:K4 (n=61), O4:KUT (n=2), and O1:KUT(n=1) (Table 1). In addition, all 64 tdh+ strains were pandemic clones with gene marker tdh+ trh- toxRS/new+. The other 9 tdh- strains belonged to serotypes O3:K6 (n=1), O6:K18 (n=1), O1:KUT (n=1), O2:KUT (n=4), O4:KUT (n=1), and O10:KUT (n=1). All 73 strains contained multivalent adhesion molecules encoding the VP1611 gene and nearly all 39 T3SS1 genes except for vopB and vscF. The 64 tdh+ strains carried all 25 T3SS2 genes, but the 9 tdh- strains were negative for the 25 T3SS2 genes (Figure 1).
-
A total of 73 V. parahaemolyticus strains were categorized into 10 STs. Four new STs (ST2894, ST2895, ST2896, and ST2897) were identified. The most frequently observed ST was ST3 (63/73, O10:K4 n=61, O3:K6 n=1, and O1:KUT n=1). The 64 pandemic strains (tdh+ trh- toxRS/new+) belonged to ST3 (O10:K4 n=61 and O1:KUT n=1) and ST2516 (O4:KUT n=2) (Table 1). All 61 O10:K4 isolates had the characteristic of tdh+ trh- toxRS/new+ orf8+ ST3, which was also characteristic of most strains from diarrhea patients.
-
The phylogenetic analysis of the 73 strains was evaluated using a WGS-SNP analysis with the reference sequence GCF_000196095.1. All of the 61 O10:K4 strains with ST3 formed the main lineage (Figure 2), which was close to the other two ST3 strains (2021VP046 belonging to O1:KUT and 2021VP010 belonging to O3:K6). The other 10 strains belonging to 5 serotypes (O6:K18, O1:KUT, O2:KUT, O4:KUT, and O10:KUT) and 9 different STs, formed the individual branches.
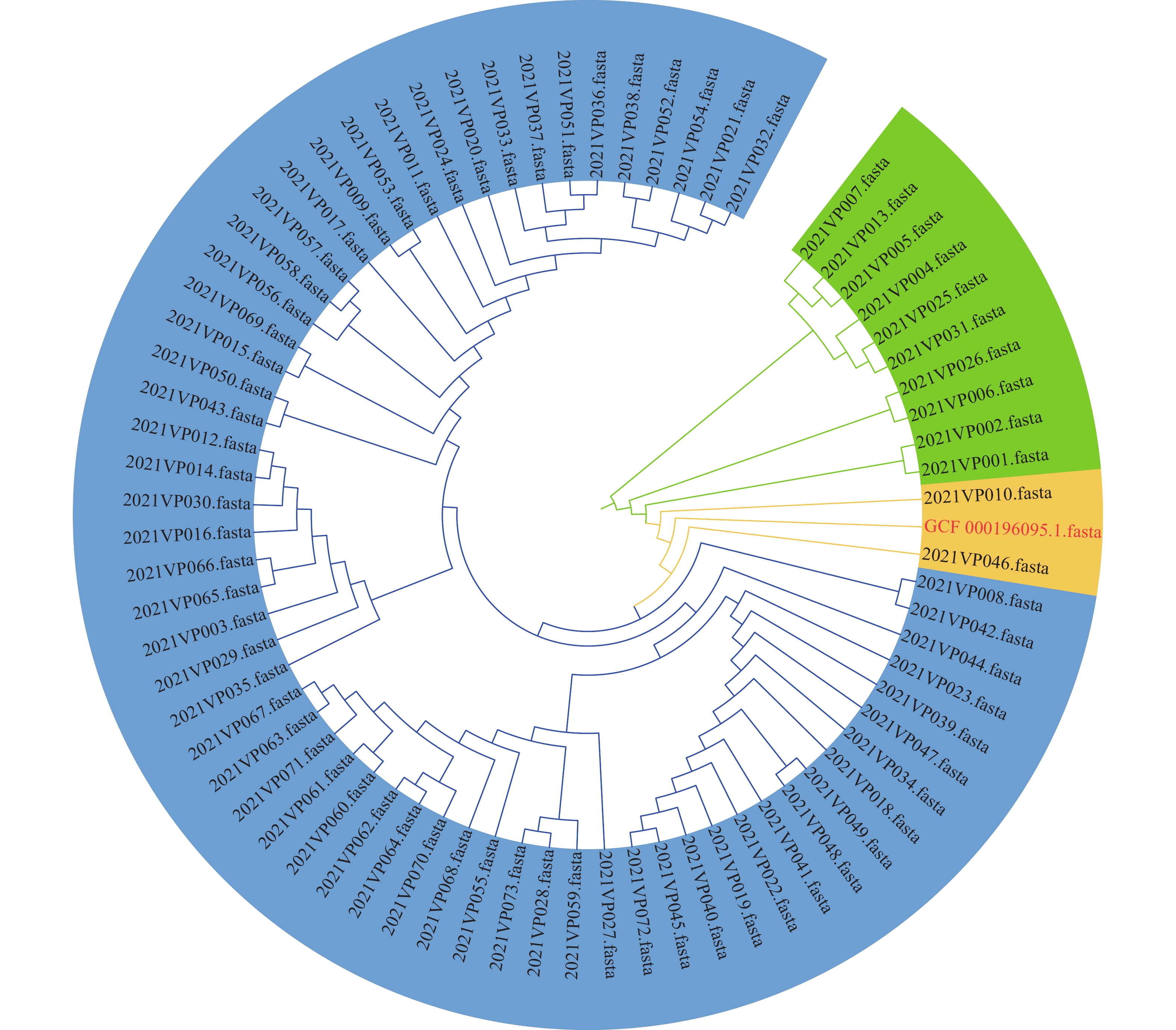 Figure 2.
Figure 2.Phylogenetic tree of 73 clinical V. parahaemolyticus strains by WGS-SNP analysis in Beijing in 2021.
Note: The 61 genomes from O10:K4 strains with ST3 were indicated within the blue ring lineage. The two genomes from the other ST3 strains (2021VP046 belonging to O1:KUT and 2021VP010 belonging to O3:K6) were indicated within the yellow ring lineage. The other 10 genomes from 5 serotypes (O6:K18, O1:KUT, O2:KUT, O4:KUT, and O10:KUT) and 9 different STs, were indicated within the green ring lineage. Abbreviations: V. parahaemolyticus=Vibrio parahaemolyticus; WGS=whole genome sequence; SNP=single nucleotide polymorphism; ST=sequence type. -
V. parahaemolyticus serotype O3:K6 with pandemic makers (tdh+ trh- toxRS/new+ orf8±) has been widespread in many countries including China since 1996 (1,4). In this study, only one of 73 clinical V. parahaemolyticus isolates was identified as O3:K6 in Beijing in 2021, which was much lower than our previous study reporting of 67.7% over the previous 10 years from 2010 to 2019 (8) and 48% of the clinical isolates of V. parahaemolyticus collected in Guangdong Province from 2007 to 2011 (9). Moreover, this O3:K6 isolate was neither a pandemic nor a pathogenic strain. Above all, 61 O10:K4 strains (83.6%) with pandemic traits (tdh+ trh- toxRS/new+ orf8+) were found for the first time and became the dominant clone instead of O3:K6 in 2021. The emergence of pathogenic and pandemic V. parahaemolyticus O10:K4 strains presented in this study should be a matter of concern for public health authorities, as the risk of outbreak rises. Recent studies have shown that at least 21 non-O3:K6 serotypes such as O4:K8, O4:KUT, and O3:K8 exhibited pandemic markers, and most likely originated from the same clones as O3:K6 (4,10). These findings suggest that the O and K antigen encoding loci are subject to exceptionally high rates of recombination (11). Serovar conversion through mutation or horizontal gene transfer of the O and K antigen encoding genes may be one means for V. parahaemolyticus to adapt to environmental changes and human immune responses (12).
In addition, this study revealed that T3SS1 genes were present in all V. parahaemolyticus strains and T3SS2 genes were predominantly present in the pathogenic and pandemic strains, indicating that the T3SS2 region may enhance virulence when present. Our results are consistent with the recent finding in an experimental animal model, which demonstrated that T3SS2 is necessary for pathogen colonization and the development of gastroenteritis (13). In this study, all the O10:K4 strains carried the entire T3SS1 and T3SS2 genes, which confirmed that the O10:K4 strains had a similar virulence as the O3:K6 pandemic clone. The 9 tdh- trh- T3SS2- strains were isolated from diarrheal patients, which also suggested that V. parahaemolyticus might harbor other virulence factors responsible for diarrhea. Therefore, the understanding of pathogenicity is still incomplete and it is necessary to discover more reliable predictors of virulence.
Among 10 STs analyzed in this study, ST3 was the most common one, which matched the findings of previous studies that ST3 was representative of pandemic clones on a global scale (14). The correlation between different serogroups and STs has been observed and some STs contained several different serogroups, such as ST3 (O1 and O3) (9,15). This phenomenon was observed in this study, that all the ST3 strains belonging to different serotypes O10:K4, O1:KUT, and O3:K6 formed a cluster, different from other STs which broke into individual branches in the phylogenetic analysis. Moreover, all the O10:K4 strains and the genetic variant O3:K6 (tdh- trh- toxRS/new+ orf8-) were placed in the same cluster, suggesting a possibility of transfer of the pandemic clone.
To the best of our knowledge, this was the first report of O10:K4 associated with diarrhea cases in China. The whole-genome sequence analysis indicated that it belonged to ST3 lineage which has the capacity to spread rapidly and the potential to replace native strains. However, it was unclear where this novel clone originated from and how it entered Beijing. Therefore, it is necessary to track the source of O10:K4 strains and to strengthen monitoring of their spread and epidemic trends through the continuous surveillance of V. parahaemolyticus in the future.
-
No conflicts of interest.
-
Staff of District CDCs of Beijing Municipality.
HTML
Study Design and Population
Detection of Bacteria and Serotyping
Antimicrobial Resistance Testing
DNA Extraction and WGS
MLST, ARGs, and VGs
Serotypes
Antibiotic Resistance Profile and Resistance Genes
Distribution of Virulence-associated Genes
MLST Analysis
Phylogenetic Analysis
| Citation: |

|