

 Download:
Download:




-
The incidence of human brucellosis is closely linked to interaction with infected animals, either through direct contact with those animals or by consuming products from infected livestock. The tracing of epidemiological sources is complicated due to the frequent movement of livestock between provinces and the variety of trade channels. From 2017 to 2022, Weihai experienced a low incidence of brucellosis, averaging 0.6365 cases per 100,000 inhabitants. During the period of June 12 to June 30, 2022, the Weihai CDC identified an outbreak comprising six brucellosis cases. The epidemiological investigation was unable to determine the precise source and route of infection. Advanced techniques such as Multiple Locus Variable-Number Tandem Repeat Analysis (MLVA) and whole genome single nucleotide polymorphism (wgSNP) analyses, utilizing data from whole genome sequencing (WGS), can offer a more definitive resolution in identifying the source of infection, potentially reducing the need for costly field investigations. Survey findings indicated that the outbreak was primarily due to live animal transport from adjacent cities and counties. Implementing novel technologies such as WGS and fostering inter-regional collaboration are essential strategies for curtailing the further transmission of brucellosis.
Whole blood samples were collected from all patients associated with the 2022 outbreak in Weihai, as well as from sporadic cases for strain isolation. The isolates underwent biochemical identification and BCSP31-polymerase chain reaction (BCSP31-PCR). Slide agglutination with monospecific anti-Brucella sera and AMOS-PCR were used for biotyping (1). Genomic DNA was extracted from the Brucella strains using the TIANamp Bacteria DNA Kit (TIANGEN, Beijing, China). The lineages and molecular typing of these strains were determined through MLVA as detailed in previous studies (2). The resulting strains were analyzed, and dendrograms were constructed with BioNumerics (version 8.0, Applied Maths, Sint-Martens-Latem, Belgium), utilizing the categorical coefficient and unweighted pair group method using the arithmetic mean (UPGMA) algorithm. WGS was performed for all strains by Novogene Bioinformatics Technology Co., Ltd, employing an Illumina NovaSeq 6000 system (PE, 150 bp reads). Brucella melitensis (B. melitensis) genomes were aligned against the reference sequence of B. melitensis bv. 1 strain 16M (RefSeq GCF_000007125.1) using CLC Genomics Workbench (version 23.0.3, Qiagen, Hilden, Germany) for SNP analysis. Data from MLVA-11, which were completely identical for strains in Shandong Province in 2022, served as the reference for Most Small Tree (MST) and SNP tree construction.
From June 12 to 30, 2022, the China Infectious Disease Surveillance and Reporting Information System detected 4 confirmed cases (patients 1–4) of brucellosis in the Huancui District of Weihai. Through active searches, 2 suspected cases (patients 5–6) were identified and later confirmed by laboratory tests. The outbreak involved a total of 6 cases: 4 cases (patients 3–6) were involved in livestock husbandry on the same farm, while 2 cases (patients 1–2) contracted the disease by consuming sheep placentas purchased from the farm. An epidemiological investigation revealed that a breeding ram bought from Rushan in August 2021 was likely the source of the infection.
In 2022, six strains were isolated in Weihai. Three strains, identified as 2022SD184, 2022SD185, and 2022SD186, were associated with patients 1, 3, and 6, respectively. The remaining strains, 2022SD060, 2022SD183, and 2022SD510, represented sporadic cases from Wendeng, Rushan, and Rongcheng, respectively. The identification of these strains as B. melitensis bv. 3 was confirmed via slide agglutination with monospecific anti-Brucella sera and AMOS-PCR. MLVA-11 analysis, which targets 11 specific Brucella loci, classified them as belonging to the East Mediterranean lineage and type 116. We expanded our molecular typing approach with additional loci for MLVA, and MST analysis using MLVA-16 data revealed the genetic distance between Weihai’s outbreak and sporadic strains (Figure 1). Two distinct MLVA-16 profiles were determined for the outbreak strains. 2022SD185 differed at one locus (bruce 09) compared to the other two strains. Furthermore, identical MLVA-16 profiles were observed among strains 2022SD184, 2022SD186, 2022SD357, and 2022SD620 from Weihai, Zibo, and Yantai cities when compared with 2022 monitoring data of Shandong Province.
 Figure 1.
Figure 1.Minimum spanning tree for Brucella melitensis generated using MLVA-16 data with Weihai isolates (green) and representative entries from monitoring data for Shandong Province in 2022.
WGS of 44 Brucella melitensis isolates from Shandong Province revealed 5,159 SNPs. A threshold of seven SNP differences was utilized to identify potentially related strain complexes (3). The analysis demonstrated that the three strains implicated in this outbreak differed by a single SNP, corroborating the epidemiological investigation outcomes and confirming their identification as an outbreak cluster. Additionally, a divergence of over 140 SNP loci was observed between the sporadic strains and the outbreak strains, indicating no evidence of transmission linkage, which aligns with the results obtained from MLVA-16 typing analyses (Figure 2). An advanced stratification analysis technique determined that the strain identified as B2022SD620, originating from Qixia City in Yantai, was identical to the strain associated with the current outbreak. By integrating epidemiological data and the geographic distribution of homologous strains, it was inferred that the spread of the epidemic likely resulted from trans-regional trade of contaminated livestock.
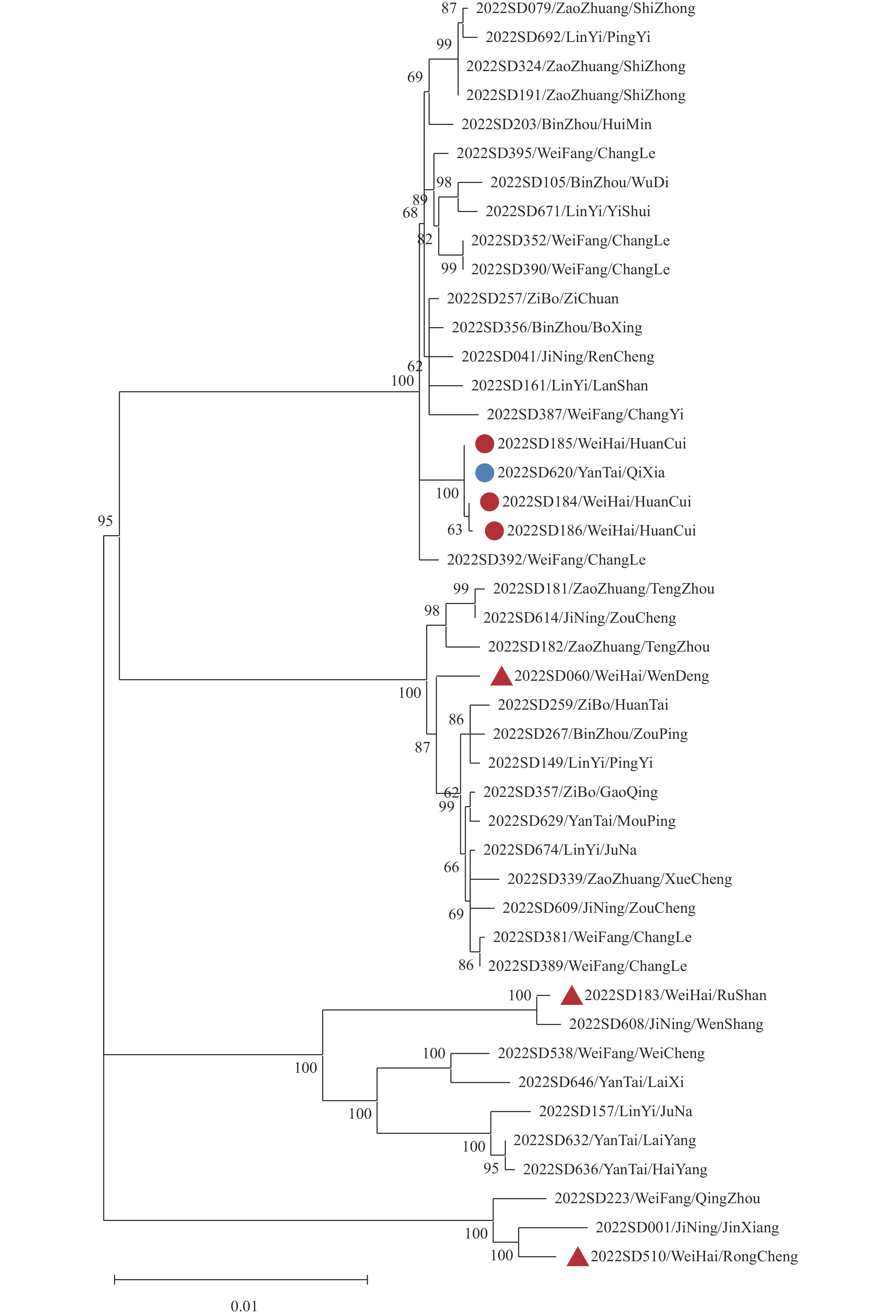 Figure 2.
Figure 2.Neighbor-joining phylogenetic tree based on Brucella melitensis wgSNP types from Shandong Province.
Note: The red dots correspond to the strain in the outbreak, and the blue dots correspond to the outbreak-associated strain from Yantai City. The red triangles represent sporadic strains from Weihai City. -
The prevalence of brucellosis in Shandong Province has seen a declining trend since its peak in 2016. However, there has been a notable resurgence of the disease since 2021. This resurgence has been largely attributed to the growth of the livestock breeding industry and increased interprovincial transport of livestock. The complexity and often clandestine nature of livestock trade, coupled with insufficient financial incentives for culling infected animals, have complicated the tracking of infection sources and destinations via epidemiological research. Our investigations revealed that in August 2021, patients had acquired sheep with unverified immunization and health statuses from Rushan, leading to subsequent infections during lambing and the handling of by-products. The substantial lapse of time between the initial outbreak and the acquisition of the infected sheep poses significant challenges to making headway in pinpointing the infection’s origins through epidemiological methods.
The classical taxonomy of B. melitensis, based on biological species classification, sorts it into three biovars, yielding limited epidemiological insight due to poor resolution among isolates. In addition, the typing identification method based on bacterial culture has a large risk of biosafety, and the judgment of experimental results has a certain subjectivity.
MLVA is considered highly effective for Brucella spp. typing, reputed for its discriminating resolution (4). Yet, the MLVA-16 method produced inconsistent results for three strains from a recent outbreak. This suggests MLVA-16’s limitation in providing precise resolution, predicting strains in long-term outbreaks, or establishing connections between strains without direct links over extended periods (3,5). To enhance the investigation’s discriminatory capability, we examined 44 genome assemblies using a wgSNP approach, setting a 7-SNP threshold to identify a cluster of related cases. This analysis revealed a strain from Yantai Penglai clustering with strains from Weihai related to the outbreak, whereas other strains sharing the same MLVA type did not. The strains from Weihai were highly homologous to those from Yantai, suggesting frequent livestock movement between the cities. Although wgSNP analysis appears to provide superior discriminatory power among available typing methods, it lacks standardized procedures and is influenced by various factors, including nucleic acid extraction, library preparation, sequencing techniques, and bioinformatics analysis methods.
In conclusion, WGS data is valuable for enhancing insights into epidemiological dynamics, especially for source tracing during outbreaks involving zoonotic transmission and illegal animal movements. WGS data is more efficient than MLVA in assessing the potential for prolonged outbreaks and accurately predicting transmission routes (6).
-
No conflicts of interest.
HTML
| Citation: |

|