

 Download:
Download:




-
Human sapovirus (HuSaV) is an enteric virus responsible for sporadic cases and outbreaks of acute gastroenteritis (AGE) globally. The prevalence of HuSaV ranges from 1% to 17% of diarrheal occurrences worldwide (1). However, the majority of studies concerning AGE outbreaks have centered on norovirus (NoV), leaving the epidemiological features of HuSaV outbreaks relatively unclear. HuSaV is a member of the Caliciviridae family, similar to human NoV, causing milder yet virtually identical symptoms, such as vomiting, diarrhea, and fever. The virus has a positive-sense, single-stranded ribonucleic acid (RNA) genome and is non-enveloped. Its approximately 7.5 kb RNA genome consists of two or three open reading frames (ORFs). SaV viruses are classified into 19 genogroups (GI to GXIX) based on the viral protein 1 (VP1) sequence and are further divided into 18 well-established genotypes: GI.1 to GI.7, GII.1 to GII.8, GIV.1, GV1, and GV.2, along with a tentative GII genotype GII.NA 1 (2-3). GI and GIV groups have been predominantly reported between October 1976 and April 2016 (4). Currently, there is a scarcity of data on the molecular epidemiology of HuSaV outbreaks in Beijing Municipality, China.
The initial identification of HuSaV in an AGE outbreak in Chaoyang District, Beijing, transpired in December 2015. Comprehending genotype distribution offers valuable information regarding transmission patterns, accurate diagnosis of circulating strains, population immunity, and potential vaccine development. Consequently, this study investigated the epidemiological trends and genetic features of HuSaV in AGE outbreaks. These findings may contribute to an enhanced understanding of HuSaV infection in Beijing.
-
This study examines AGE outbreaks from January 2015 to December 2021 in Chaoyang District of Beijing. AGE cases were defined as patients exhibiting diarrhea (three or more loose stools within a 24-hour period) and/or vomiting (one or more episodes). An outbreak was characterized by the occurrence of three or more epidemiologically linked AGE cases within a 3-day period. Outbreaks were reported to community health centers by schools, kindergartens, and other institutions, and subsequently to the Chaoyang District CDC.
The Chaoyang District CDC conducted investigations within 24 hours of receiving a report, and fecal specimens or anal swabs were collected from cases within 48 hours of initiating the investigation to detect pathogens. An AGE outbreak surveillance network is already established in Beijing (5). In the Chaoyang CDC, AGE outbreak diagnostic tests include detection of NoV, sapovirus (SaV), rotavirus (RV), astrovirus (AstV), and enteric adenovirus (AdV). A HuSaV outbreak was confirmed if two or more AGE cases tested positive for HuSaV via real-time polymerase chain reaction (PCR).
-
HuSaV-positive samples were amplified using primers SLV531/SLV574, yielding a 434-bp PCR product. PCR products were purified and sequenced. All sequences were aligned using BioEdit (version 7.0.5.3, Borland, Scotts Valley, USA). Nucleotide sequences from this study and reference strains were compared to examine their differences using the MegAlign module of the Lasergene software package (version 7.1, DNASTAR, Madison, WI). The number of single nucleotide polymorphism (SNP) sites was calculated based on the alignments. Fisher’s exact test was employed to evaluate the significance of frequency differences among the three groups of bases. P-values were corrected using the Benjamini-Hochberg (BH) method. Genotypes were determined through phylogenetic analysis, utilizing the neighbor-joining method and a bootstrap test with 1,000 iterations, implemented in MEGA software (version 6.0, Mega Limited, Auckland, New Zealand). Reference strains for different HuSaV genotypes were selected and obtained from the GenBank database.
-
In a review of 678 AGE outbreaks from 2015 to 2021 in Chaoyang District, 71 (10.5%) were laboratory-confirmed as HuSaV-associated. HuSaV ranked second among the five most common diarrhea-associated viruses, including NoV (59.3%), SaV (10.5%), AstV (2.4%), enteric AdV (0.9%) , and RV (0.2%). Furthermore, no pathogens were detected in 26.7% of the samples. Of the HuSaV-associated cases, 67 (94.4%) had single HuSaV infections, two had co-infections with AstV, one had a co-infection with AdV, and one had a triple infection, which involved GII NoV and AstV.
HuSaV predominantly infected children under 5 years of age in kindergarten settings among the enrolled subjects. Out of the 71 HuSaV outbreaks, 90.1% (64/71) occurred in kindergartens, while 9.9% (7/71) took place in primary schools. The median age of the HuSaV-positive individuals was 4 years (range: 2–10 years). The highest proportion of infected individuals was found in the 4-year-old age group (39.0%), followed by the 3-year-old (28.6%) and 5-year-old (18.2%) age groups. The proportion of HuSaV-positive individuals in the under-5-years group was significantly higher than that in the older-than-5-years group (P<0.001, Chi-square test).
The modes of transmission were identified for 63 outbreaks. The predominant mode of transmission was person-to-person, accounting for 62 of the 63 outbreaks (98.41%). Water-borne transmission was the next most common, contributing to one of the 63 outbreaks (1.59%).
HuSaV-caused AGE outbreaks occurred every year, and the constituent ratio ranged from 5.9% to 15.0%. Outbreaks typically occurred in two periods: May to June and September to December, with the exception of 2020 (Table 1). A few cases occurred in March and April, while no cases were reported in January, February, July, and August.
Genotype Proportion of outbreaks (%) 2015 (n=2) 2016 (n=3) 2017 (n=18) 2018 (n=18) 2019 (n=16) 2020 (n=1) 2021 (n=26) Total (n=71) GI.1 − 33.3 16.7 20.0 − 100 11.5 12.7 GI.2 − − 11.1 20.0 6.3 − 11.5 9.9 GI.5 − − − − 12.5 − − 2.8 GI.6 − − − − − − 3.9 1.4 GII.1 − − 5.6 − − − − 1.4 GII.3 100 33.3 44.4 40.0 62.5 − 61.5 54.9 GII.5 − 33.3 − − 18.8 − 3.9 7.0 Untyped − − 22.2 20.0 − − 7.7 9.9 Note: “−” means that no related outbreak was detected.
Abbreviation: HuSaV=human sapovirus.Table 1. Number and proportion of HuSaV outbreaks by genotype and by year.
-
The genetic diversity of AGE-causing HuSaVs was analyzed in this study. Out of 71 HuSaV outbreaks, the partial VP1 gene was successfully amplified and sequenced in 64 outbreak samples. Seven genotypes were identified, with 19 (26.8%) belonging to the GI genogroup, 45 (63.4%) to the GII genogroup, and 9.9% remaining untyped. The specimens from these untyped outbreaks had low viral loads and could not be sequenced. HuSaV outbreaks caused by the GII genogroup were predominant in all reported settings, including 40 outbreaks in kindergartens and 5 outbreaks in primary schools. In contrast, all GI genogroup outbreaks occurred exclusively in kindergartens. No significant differences were observed in the gender of infected individuals, month of infection, or median age between HuSaV GI and GII outbreaks.
Furthermore, it is worth noting that GII.3 emerged as the most predominant genotype, accounting for 54.9% of the cases and being detected in almost every month. Other genotypes observed included GI.1 (12.7%), GI.2 (9.9%), GII.5 (7.0%), GI.5 (2.8%), GI.6 (1.4%), GII.1 (1.4%), and untyped (9.9%). Additionally, the composition of genetic diversity exhibited variance across different years (Table 2). However, no instances of multiplex HuSaV infections involving distinct genotypes within a single outbreak were identified in this study.
Time 2015 2016 2017 2018 2019 2020 2021 Total January − − − − − − − − February − − − − − − − − March − − GII.3 (1), GI.2 (1) − GII.5 (1) − − GII.3 (1), GII.5 (1), GI.2 (1) April − − − − − − GI.1 (1), GI.2 (1) GI.1 (1), GI.2 (1) May − GII.3 (1) − − − − GII.3 (6), GI.1 (1), GI.2 (1), GI.6 (1), GII.5 (1) GII.3 (7), GI.1 (1), GI.2 (1), GI.6 (1), GII.5 (1) June − − GI.1 (1) GII.3 (1), untyped (1) GII.3 (1), GI.2 (1) − GII.3 (7), GI.1 (1), GI.2 (1), untyped (1) GII.3 (8), GI.1 (2), GI.2 (2), untyped (2) July − − GII.3 (1) − − − GII. 3(1), untyped (1) GII.3 (2), untyped (1) August − − − − − − − − September − GII.5 (1) GII.3 (3), GI.1 (1), untyped (1) − GII.3 (3) − GII.3 (1) GII.3 (8), GII.5 (1), GI.1 (1), untyped (1) October − GI.1 (1) GII.3 (2), GI.1 (1), GII.1 (1) − GII.3 (1), GI.6 (1) − − GII.3 (3), GI.1 (2), GII.1 (1), GI.6 (1) November GII.3 (2) − GI.2 (1) GII.3 (1), GI.1 (1), GI.2 (1) GII.3 (3), GII.5 (1) GI.1 (1) GII.3 (1) GII.3 (7), GII.5 (1), GI.1 (2), GI.2 (2) December − − Untyped (3) − GII.3 (2), GI.5 (1), GII.5 (1) − − GII.3 (2), GI.5 (1), GII.5 (1), untyped (3) Note: The numbers in parentheses represent the number of samples classified to the corresponding genotype.
“−”means that no related outbreak was detected.
Abbreviation: HuSaV=human sapovirus; AGE=acute gastroenteritis.Table 2. Monthly distribution of HuSaV AGE outbreaks in Chaoyang District of Beijing, from January 2015 to December 2021.
-
In order to determine the genetic relationships of the partial VP1 genes of the HuSaV detected in this study with those previously reported, phylogenetic trees were constructed and analyzed (Figure 1). Among the strains detected in this study, nine strains clustered with GI.1 reference sequences from Japan, China, the Republic of Korea, and the Republic of Tunisia, exhibiting sequence similarity ranges of 99.0%–100%, 98.8%–99.8%, 97.4%–98.6%, and 96.6%–97.8%, respectively. Seven strains clustered with GI.2 sequences from China and French Guiana, with similarity ranges of 99.5%–100% and 97.4%–97.6%, respectively. Seven strains clustered with GI.5 sequences from Japan, displaying a similarity of 98.3%. Two strains clustered with GI.6 sequences from Japan and the United States, with a similarity of 99.5%. One strain clustered with GII.1 sequences from Malaysia and China, with similarities of 94.0% and 94.2%. Thirty-nine strains clustered with GII.3 sequences from Russia, with similarity ranging from 96.4%–98.6%. Five strains clustered with GII.5 sequences from the Republic of Guatemala and Taiwan, China, with similarities of 95.4%–96.2% and 93.0%–93.5%, respectively.
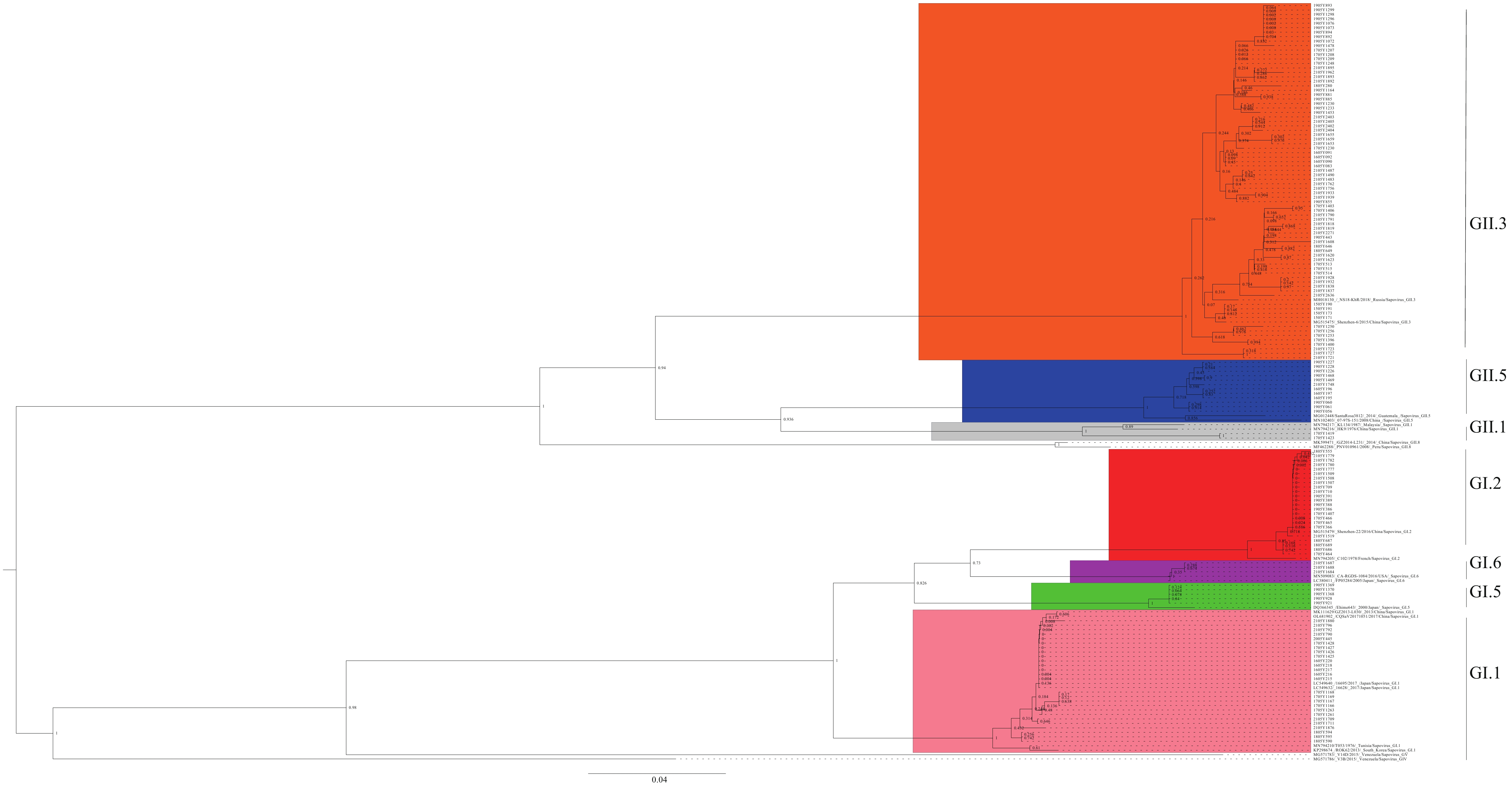 Figure 1.
Figure 1.Phylogenetic analysis of HuSaV based on partial VP1 nucleotide sequences.
Note: The phylogenetic tree was constructed using the neighbor-joining method and a bootstrap test with 1,000 iterations.
Abbreviation: HuSaV=human sapovirus; VP1=viral protein 1.
As previously mentioned, GII.3 was responsible for 86.7% (39/45) of HuSaV outbreaks within the GII genogroup. A phylogenetic tree was constructed using only the GII.3 HuSaV isolates from this study, revealing that the 39 GII.3 outbreak strains could be categorized into three distinct groups: I, II, and III. These groupings were further supported by SNP analysis, which identified a total of 15 SNPs with significant differences among the three groups (Figure 2). Of the 39 GII.3 outbreaks analyzed, 12 (30.8%) were attributed to Group I, 1 (2.6%) to Group II, and 26 (66.7%) to Group III.
 Figure 2.
Figure 2.Phylogenetic and SNP analysis of partial HuSaV VP1 genes from all GII.3 isolates in the present study.
Abbreviation: HuSaV=human sapovirus; VP1=viral protein 1; SNP=single nucleotide polymorphism. -
Viral infections continue to be a significant cause of AGE in children globally. The extensive viral diversity presents a substantial challenge for the development of effective vaccines. Currently, only RV vaccines have been commercialized for worldwide use. Consequently, monitoring the genetic diversity of AGE-associated viruses, particularly uncommon or neglected ones such as HuSaV, is crucial. The findings from this study indicate that HuSaV was the second most prevalent cause of AGE outbreaks, following NoV, in the Chaoyang District of Beijing, China, between 2015 and 2021, which is consistent with observations in some other countries (6-7). Nonetheless, the genetic diversity of HuSaV found in this study differs from previous reports in both China (8-9) and other countries (10-13), where the GI genogroup was predominant.
The predominant genotype in Chaoyang District is GII.3, which contrasts with the previously reported genotype GI in both China and other countries. Our study documents an increase in HuSaV outbreaks attributed to genogroup II, particularly GII.3, for the first time, as compared to sporadic occurrences in other regions of China (14-16). Consequently, we hypothesize that GII.3 HuSaV underwent a transition from quantitative to qualitative change due to rapid evolution in China. This assumption is supported by evolutionary dynamics analysis, which suggests that the less common GII.3 HuSaVs evolved at a faster rate than the predominant GI.1 HuSaVs (16).
The epidemic seasons in Chaoyang District occur from May to June and from September to December, which deviate from those observed in southern China. In contrast to other reports on the seasonal distribution of HuSaVs, outbreaks in Beijing typically take place during two periods, as depicted in Figure 1. This variation significantly differs from the early spring occurrences reported in southern China (9), which may result from the considerable difference in latitudes between the two regions. In Japan, researchers have also discovered that the detection rate of HuSaV within a month varied across different years (6). As attention towards HuSaV increases and detection methods improve, fluctuations in the distribution patterns of HuSaVs from place to place and year to year could change. Consequently, the factors contributing to these variations may become more evident in future studies.
There were several limitations observed in our study. First, insufficient data and resources were available to establish a relationship between SNPs and epidemiological features among different groups of GII.3 HuSaVs. Second, due to the emergence of coronavirus disease 2019, only one HuSaVs outbreak was observed in 2020, resulting in a discontinuity in our study’s data.
In conclusion, HuSaV is a primary cause of diarrhea, with GII.3 emerging as the predominant strain, potentially leading to an increase in cases and outbreaks of diarrhea in the near future. Continuous surveillance and timely data updates on HuSaV within China are essential to inform adjustments to prevention and control strategies against HuSaV-induced AGE during this potential transition period.
HTML
Data Sources
Sequencing and Data Analysis
Detection and Epidemical Distribution of HuSaV
Distribution of HuSaV Genotypes
Phylogenetic Analysis of HuSaV Outbreaks
| Citation: |

|