

 Download:
Download:




-
Prion disease (PrD) is a group of fatal and transmissible spongiform encephalopathies (TSEs) affecting humans and species of animals. Human PrDs are classified into sporadic, genetic, and acquired forms. More than 85% of all PrDs were sporadic Creutzfeldt-Jacob disease (sCJD). 10%–15% of PrDs are predominantly inherited involving in different mutations in prion protein (PRNP) gene, including genetic CJD (gCJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and fatal familial insomnia (FFI). Less than 1% of PrDs were acquired, and the majority of patients had definite iatrogenic histories (iatrogenic CJD, iCJD) (1). Since the outbreak of bovine spongiform encephalopathy (BSE) in cattle in the UK and other countries in the 1980s, a new form of human PrD (variant CJD, vCJD) emerged, caused by consuming food contaminated with the BSE agent.
Human PrD or CJD was rarely recognized and diagnosed in China till the end of the 1980s. Prof. Tao Hung from Chinese Academy of Preventive Medicine, Prof. Shihe Lin from Bethune Medical University, and Prof. Yupu Guo from Peking Union Hospital are the representative pioneers in the field of prion study in China. A collaborating network was set up in the 1990s among several hospitals and research units. After development of the essential laboratory tools for CJD diagnosis, a diagnostic center for human PrD was established in 1999 in the Institute of Virology, Chinese Academy of Preventive Medicine. In 2002, the first surveillance program for PrD/CJD was launched in Beijing Municipality, Xi’an City, Guangzhou City, and Changchun City. In 2006, the national surveillance program was officially conducted under the leadership of China CDC, which consisted of the university hospitals and provincial CDCs in 10 provincial-level administrative divisions (PLADs) at the beginning (2-3) and gradually extended to almost all provinces in the mainland of China now. Meanwhile, Chinese national surveillance for PrD/CJD joined the international surveillance network under the umbrella of the World Health Organization (WHO), e.g., Surveillance for vCJD in Central and Eastern Europe and China.
-
Human PrD or CJD was rarely recognized and diagnosed in China till the end of the 1980s. Till now, Chinese surveillance network for PrD consists of 1 national center, 12 provincial units, 15 consultant hospitals (3-4), and gradually extended to almost all provinces in the mainland of China now. The case referring, the data feedback and follow-up survey were conducted according to the surveillance technique documents. The laboratory tests were performed, including routine neuropathology, immunohistochemistry and Western blot for scrapie-like prion protein (PrPSc) in brains, Western blot for cerebrospinal fluid (CSF) 14-3-3, enzyme-linked immunosorbent assay (ELISA) for CSF tau, prion protein gene (PRNP) PCR and sequencing. Recently, real-time quaking-induced conversion (RT-QuIC) was also applied to the specimens of CSF and skin. The suspected PrD/CJD cases under the national PrD surveillance were diagnosed and subtyped based on the surveillance document issued by China CDC and the diagnostic criteria for CJD issued by the National Health Commission. By the end of 2021, 5,078 suspected CJD cases were reported to the national center, among them 1,900 were sCJD and 243 were different types of genetic PrDs (gPrDs). No iCJD or vCJD cases were identified (Table 1).
Year Referred sCJD gPrD Annual total gPrD Annual total PrD gCJD FFI GSS 2006 80 20 1 2 0 3 23 2007 113 31 3 0 0 3 34 2008 102 33 1 2 1 4 37 2009 164 32 3 3 0 6 38 2010 171 47 5 3 1 9 56 2011 184 57 5 2 1 8 65 2012 242 64 8 5 0 13 77 2013 299 116 9 3 0 12 128 2014 324 143 8 8 2 18 161 2015 366 135 17 4 1 22 157 2016 449 159 16 5 3 24 183 2017 504 225 20 10 3 33 258 2018 537 214 16 5 2 23 237 2019 520 189 25 5 1 31 220 2020 458 179 18 0 1 19 198 2021 549 256 12 1 2 15 271 Total 5,078 1,900 167 58 15 243 2,143 Abbreviation: sCJD=sporadic Creutzfeldt-Jacob disease; gPrD=genetic prion disease; gCJD=genetic CJD; FFI=fatal familial insomnia; GSS=Gerstmann-Sträussler-Scheinker syndrome. Table 1. Annual numbers of the referred, sCJD and gPrD cases from 2006 to 2021.
-
Based on the previously published data (2-6) and the data of the last five years, the onset ages of sCJD patients ranged from 19 to 86 years with the median of 62 years. More cases (roughly 40%) were in the group of 60–69 years. The gender ratio was 1.06∶1 (Males∶Females). Except Xizang Autonomous Region (Tibet), sCJD cases were identified in all PLADs in the mainland of China. There was no geographic, seasonal or occupational association.
The initial symptoms of sCJD cases varied largely. Progressive dementia was most frequently recorded (about 41%), followed by cerebellum and visual disturbances (18%), mental problems (13%), and pyramidal and extrapyramidal symptoms (10%). More neurological abnormalities gradually displayed along with disease progression. Dementia was noted in all sCJD cases. The other four sCJD associated symptoms were also frequently recorded, i.e., visual or cerebellar disturbance (67%), myoclonus (76%), pyramidal or extrapyramidal symptoms (80%), and mutism (39%). The portions of the patients having dementia plus 4, 3, and 2 other neurological symptoms were 19%, 40%, and 41%, respectively.
Periodic sharp wave complexes (PSWC) on EEG were recorded in roughly 50% sCJD patients. Abnormalities on MRI (symmetrical or asymmetrical cortical “ribbon” signs on diffusion weighted imaging DWI, a high signal in the caudate/putamen, or a high signal in the bilateral posterior tuberosity of the thalamus in the proton the density phase) were reported in 68% sCJD cases. 77% sCJD cases showed CSF 14-3-3 positive during the clinical course. Increased CSF tau levels (>1,400 pg/mL) were also observed in 87% of the tested cases. Neuropathological and molecular assays of PrPSc in the brain tissues, either postmortem or biopsy, from a small number of sCJD patients showed widely distribution of small granules in brain tissues and type-1 PrPSc molecule.
The majority of sCJD patients progressed rapidly. The clinical durations varied from 2 to 24 months after onset, with the median survival of 5.3 months. Analysis of the durations with other important factors did not show a significant association, including the onset ages, genders, occupations, personal economic situations, clinical symptoms, abnormalities in clinical examinations and laboratory tests.
-
The total Chinese gPrD cases accounted for 11.1% of all diagnosed PrDs, including gCJD, FFI, and GSS (7-8). Genetically, 19 different mutations in PRNP were identified in Chinese gPrDs (Figure 1). The most frequent mutation was T188K (9-10), which accounted for 29.8% of all gPrD cases. The mutations with more than 10 cases were D178N (25.7%) (11-13), E200K (18.8%) (14–15), E196A (7.3%) (16–17) and P102L (6.4%) (18-19). Other mutants with less than 10 cases were E196K (20), V203I (21), R208H (22), V210I, G114V (23-24), R148H, P105L, V180I (25), T183A, and E200G. Four cases were confirmed to contain mutations in the octapeptide repeat (OR) region: one with 7 extra ORs (26), one with 1 extra OR, one with 1 OR deletion (27), and one with 1 octarepeat deletion together with a G114V point mutation in the same PRNP allele. Such pattern of mutations in gPrDs was not only completely different from Caucasian cases in European and North American countries, but also different from Japanese and the Republic of Korean cases.
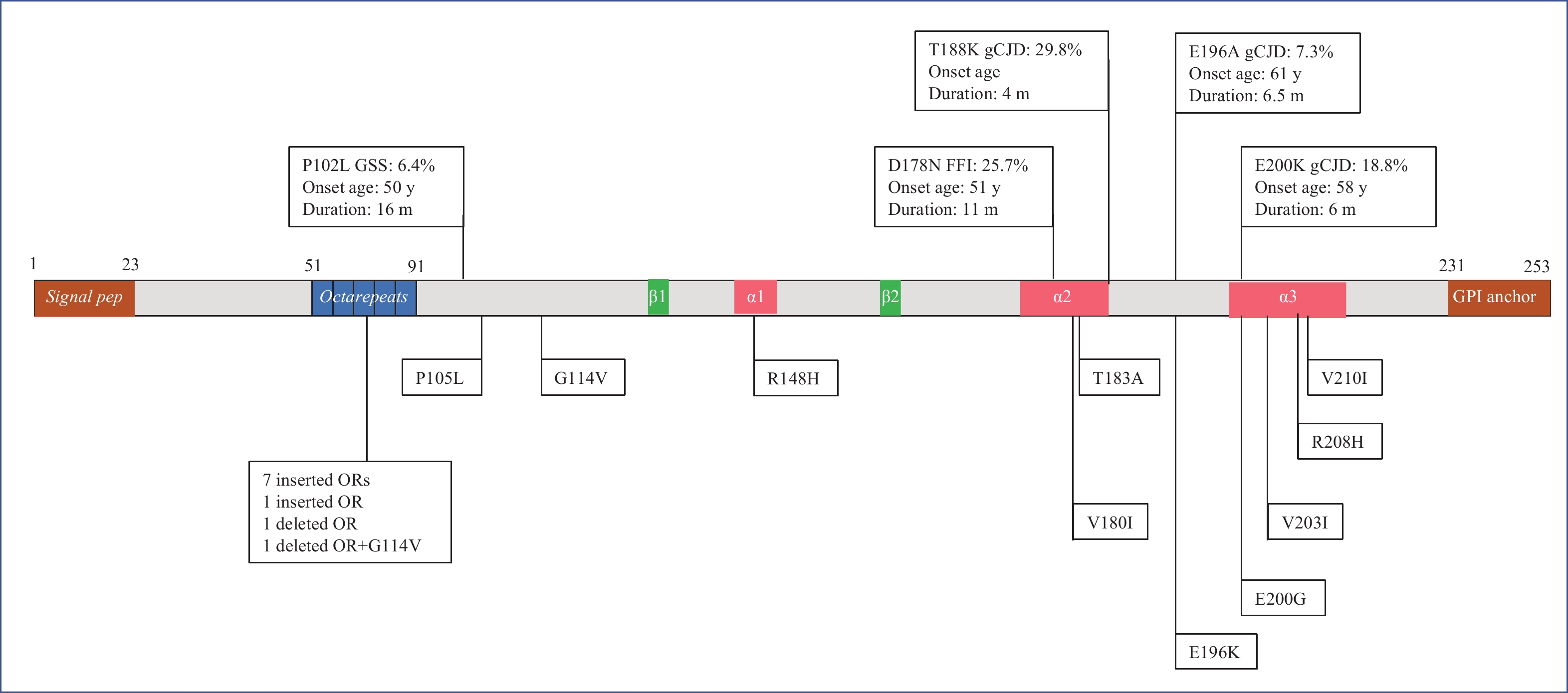 Figure 1.
Figure 1.Schematic structure of PrP protein and 19 different genotypes of Chinese gPrDs.
Note: The signal peptide at N-terminus, GPI anchor at C-terminus, five OR, three α-helix regions, two β-sheet regions are shown inside. The serial numbers of amino acid are indicated above the schematic structure. The top five frequently observed gPrDs are illustrated in the upper part, with their proportion (%), the median of onset age (year, y), the median of duration (month, m). The rest of mutants are shown in the lower part.
Abbreviation: OR=octapeptide repeat; gCJD=genetic Creutzfeldt-Jacob disease; PrP=prion protein; gPrDs=genetic prion diseases; GPI=glycosylphosphatidylinositol; GSS=Gerstmann-Sträussler-Scheinker syndrome.
The onset ages of Chinese patients with gPrDs were generally younger than that of sCJD patients, with the median of 50 years old (19,85). Different gPrDs showed obvious diversity in their onset age. The median onset ages of P102L GSS (50 years) and D178N (51 years) FFI were younger than those of T188K (61 years), E196A (61 years) and E200K (57 years) gCJD. 78% of P102L GSS and 54% of D178N FFI cases had a definite family history, whereas only approximately 15% of T188K and E200K gCJD patients recorded family history. Unlike sCJD, some types of gPrDs showed geographic association, e.g., more D178N FFI cases in Henan and Guangdong, while more cases of E200K cases in the northern provinces.
Besides the great differences in clinical manifestations between gCJD, GSS, and FFI, the profiles of EEG, MRI, CSF 14-3-3, and CSF tau among the gPrD patients with different mutations were also different. PSWC on EEG was identified in 48% E200K gCJD cases, but low in E196A (25%), T188K (27%), P102L (20%), and extremely rare in D178N (2%). Special abnormalities on MRI were frequently recorded in most types of gPrDs, i.e., P102L (92%), T188K (78%), E196A (73%), E200K (85%), but infrequently in D178N (25%). Positive CSF 14-3-3 was detected more often in gCJD patients (62% in T188K, 75% in E196A, 71% in E200K), but less in D178L FFI (39%) and P102L GSS (41%) cases. Increased CSF tau was observed in a small portion of P102L (24%), but frequently in D178N (59%), T188K (54%), E196A (80%), and E200K (68%).
The survival time of P102L cases was notably longer than that of the others, with a median (50% percentile) of 16 months. 60% of P102L cases survived longer than 12 months and 40% longer than 24 months. The median survival time of D178N cases was 11 months, among them 30% cases survived longer than 1 y, whereas those of T188K (4 months), E196A (6.5 months), and E200K (6 months) were clearly shorter. The ratios of the cases alive longer than 1 y after onset were 6.5% in T188K, 17% in E196A, 20% in E200K, respectively. Taken together, the gPrD cases showed an obvious difference in the characteristics of epidemiology, clinic and laboratory compared to sCJD cases.
-
Two polymorphisms in PRNP, codon 129 and codon 219, affect the susceptibility and phenotype of PrDs. Previous studies have confirmed that East Asians have predominant genotype of M129M (92%–95%) compared to Caucasians (50%–70%). PRNP sequencing of more than 5,000 referred cases in the national surveillance for PrD also proposed absolutely predominant patterns of M129M and homozygote of glutamic acid at codon 219 (E219E). Compared to the group of non-CJD, both sCJD and gPrD cases showed even higher ratios of M129M and E219E (Table 2).
Disease Codon 129 Codon 219 M129M M129V V129V E219E E219K K219K sCJD 98.5 1.5 0 98.9 1.1 0 gPrD 98.2 1.8 0 98.7 1.3 0 non-CJD 96.8 3.2 0 93.5 6.5 0 Referred 97.6 2.4 0 96.3 3.7 0 Abbreviation: PRNP=prion protein gene; sCJD=sporadic Creutzfeldt-Jacob disease; gPrD=genetic prion disease. Table 2. Proportions of the polymorphism at codon 129 and 219 in PRNP in different diseases from 2006 to 2021 (%).
-
One of the most significant achievements of Chinese national PrD surveillance is comprehensive description of the epidemiological, clinical and laboratory features of Chinese PrDs. It enriches the overall understanding of human PrDs. sCJD is the predominant type of PrDs in China, showing similar characteristics to those of other countries. About 10% PrDs cases are gPrDs consisting of 19 different genotypes. Notably, the profile of Chinese gPrDs is identical not only to that of Western countries but also to our neighboring ones, i.e., Japan and the Republic of Korea. T188K and E196A gCJDs are popular in China, but extremely rare in other countries. On the other side, some frequently reported gPrDs in Japanese, such as P105L GSS and M232V gCJD, are rare in Chinese. It verifies again the ethno-correlation of distributions of PRNP variants. There is no vCJD case in the mainland of China, which may, from another angle, reflect a BSE-free environment.
Our surveillance for human PrDs promotes the recognition and diagnosis of such rare diseases. Along with the implementation of PrDs surveillance, more and more hospitals from various provinces in the mainland of China referred CJD cases and the annual case numbers referred to the surveillance center were over five hundred in the past five years. Our PrDs surveillance also proposes strong technique support for diagnosis and differential diagnosis of PrDs for hundreds of hospitals over China. Thousands of laboratory tests of clinical samples each year, countless communications and consultants with physicians, timely feedback of the diagnosis and disposal suggestion for each referred case under the national PrDs surveillance set up a good example for the combination of clinical and preventive medicine. Our surveillance program also functions as a platform to communicate with the family members and relatives of PrD patients, providing medical consultant and daily care suggestions and greatly alleviating the fear of this unfamiliar disease due to its infectious potential.
As a neurodegenerative disease, there is still a lack of specific therapeutic or prophylactic tools for any type of PrD. The wide distribution of infectious prions in the central nerve system, eyeballs, and some other peripheral lymph tissues has caused great concern about iatrogenic infection, e.g., neurosurgical operation, organ transplantation, and even blood transfusion. Moreover, the impacts and threats of BSE and other animal prion diseases, such as chronic wasting disease in Cervidae, on public health of humans are still long-standing. Long-term surveillance for human and animal PrDs is still needed.
HTML
| Citation: |

|